

Prophage-encoded antibiotic resistance genes are enriched in human-impacted environments
- PMID: 39333115
- PMCID: PMC11437078
- DOI: 10.1038/s41467-024-52450-y
Prophage-encoded antibiotic resistance genes are enriched in human-impacted environments
Abstract
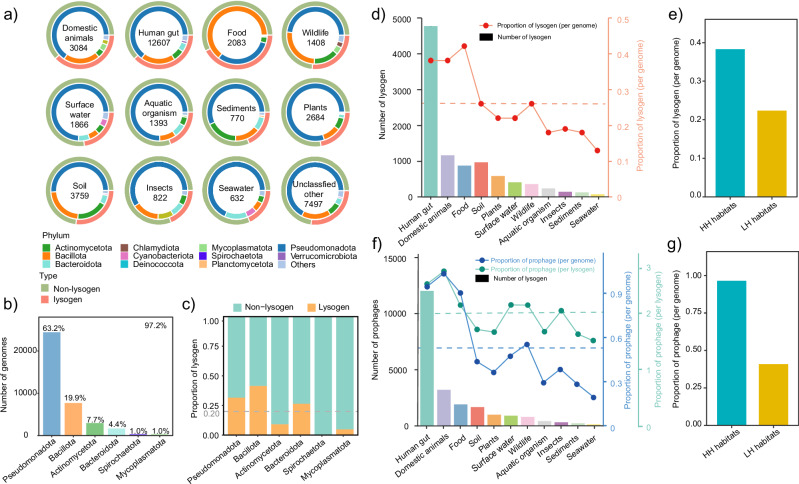
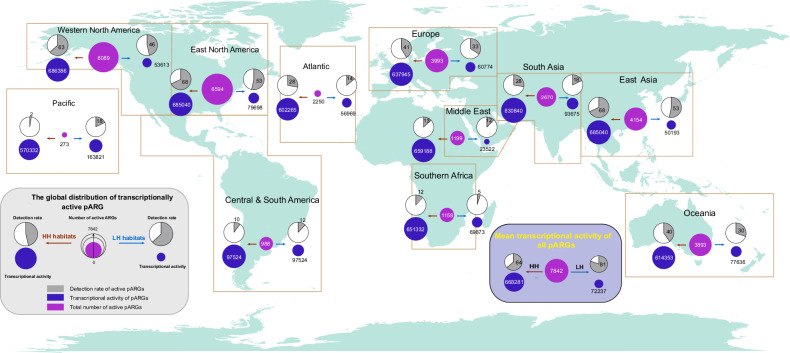
The spread of antibiotic resistance genes (ARGs) poses a substantial threat to human health. Phage-mediated transduction could exacerbate ARG transmission. While several case studies exist, it is yet unclear to what extent phages encode and mobilize ARGs at the global scale and whether human impacts play a role in this across different habitats. Here, we combine 38,605 bacterial genomes, 1432 metagenomes, and 1186 metatranscriptomes across 12 contrasting habitats to explore the distribution of prophages and their cargo ARGs in natural and human-impacted environments. Worldwide, we observe a significant increase in the abundance, diversity, and activity of prophage-encoded ARGs in human-impacted habitats linked with relatively higher risk of past antibiotic exposure. This effect was driven by phage-encoded cargo ARGs that could be mobilized to provide increased resistance in heterologous E. coli host for a subset of analyzed strains. Our findings suggest that human activities have altered bacteria-phage interactions, enriching ARGs in prophages and making ARGs more mobile across habitats globally.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
