

CNVoyant a machine learning framework for accurate and explainable copy number variant classification
- PMID: 39333267
- PMCID: PMC11437066
- DOI: 10.1038/s41598-024-72470-4
CNVoyant a machine learning framework for accurate and explainable copy number variant classification
Abstract
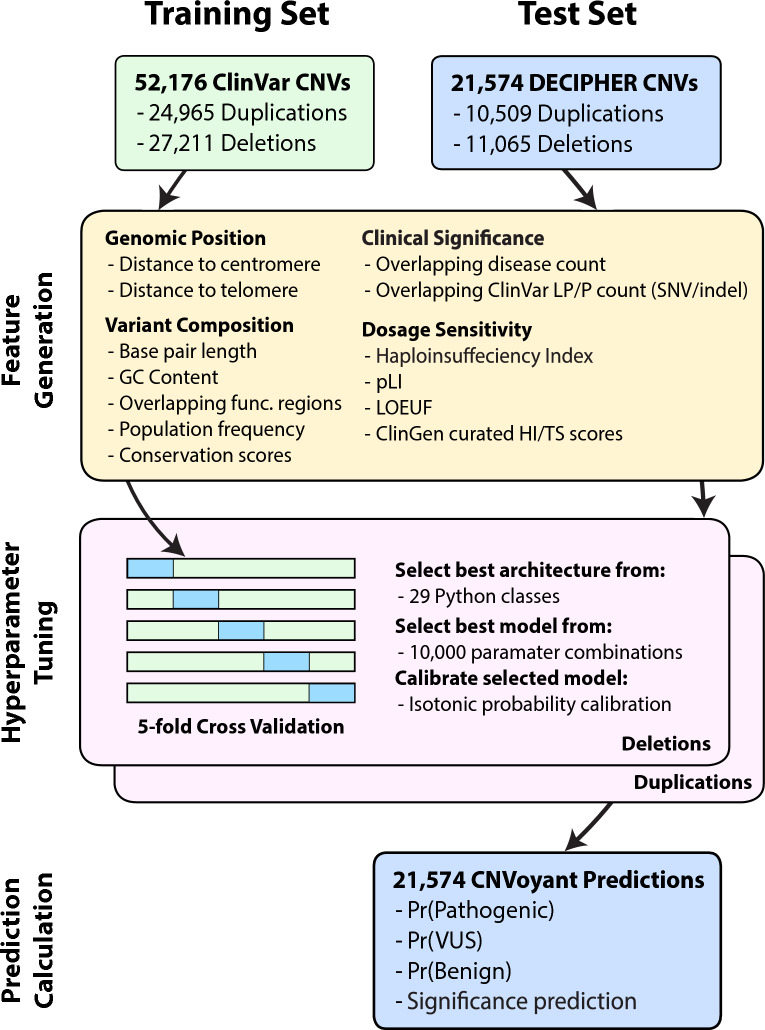
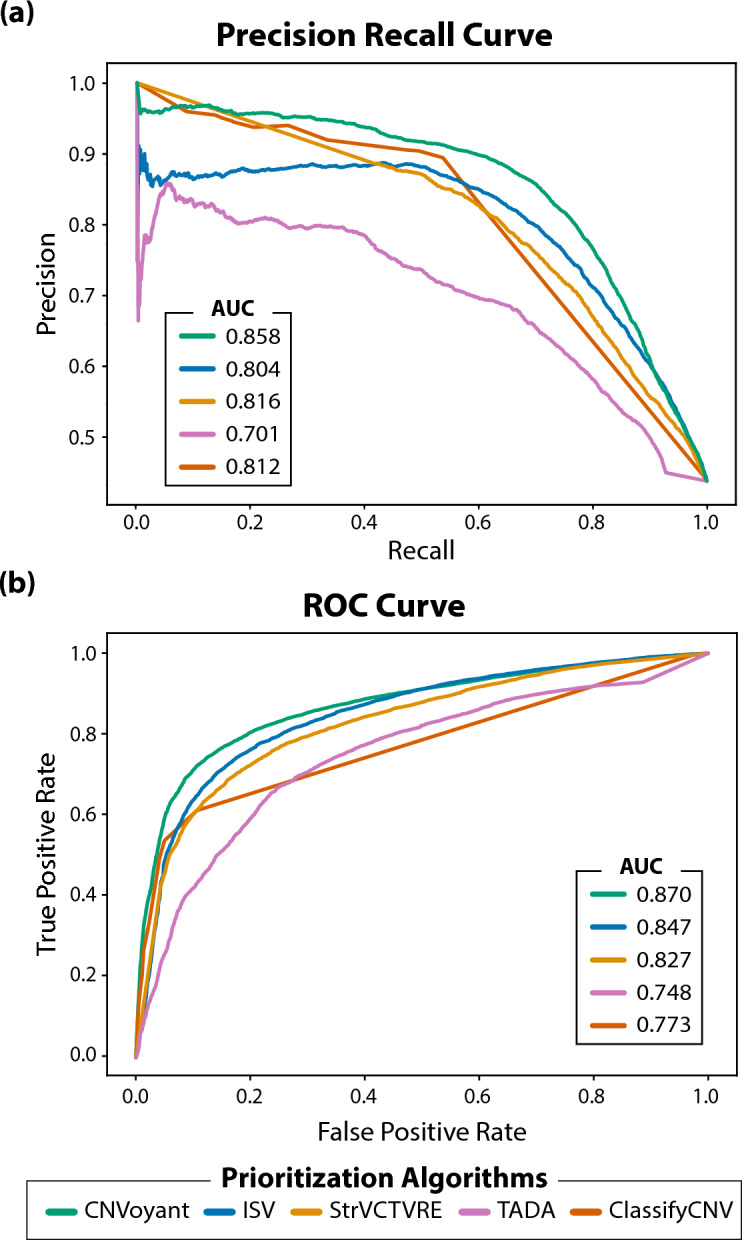
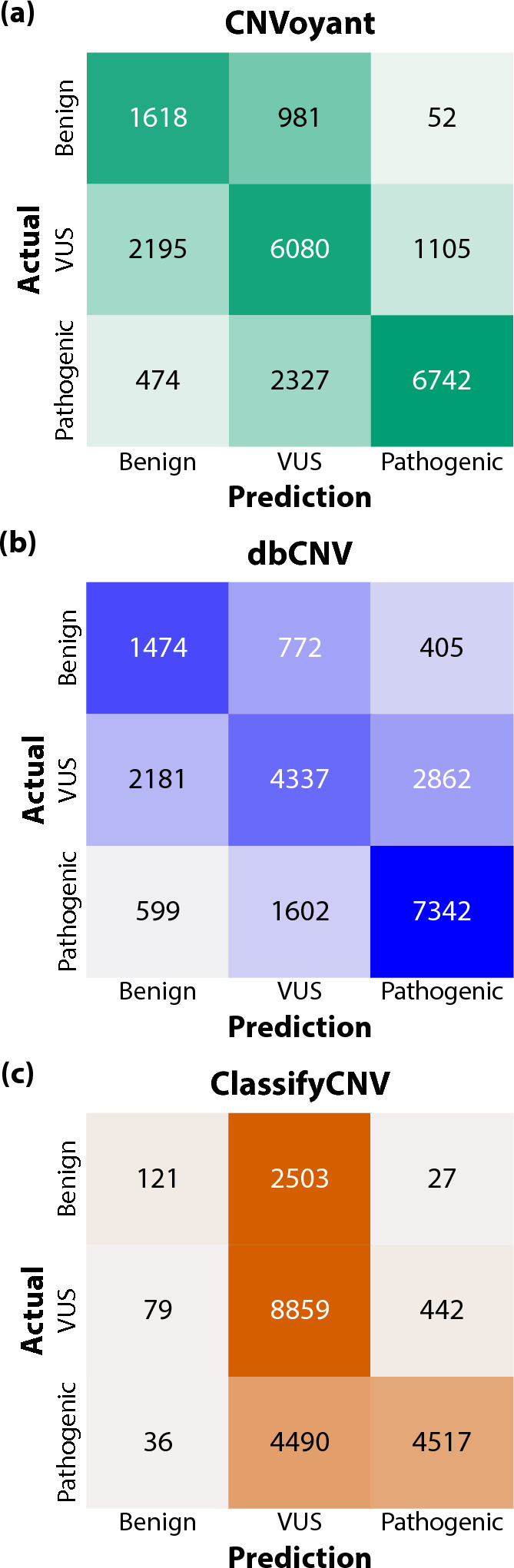
The precise classification of copy number variants (CNVs) presents a significant challenge in genomic medicine, primarily due to the complex nature of CNVs and their diverse impact on rare genetic diseases (RGDs). This complexity is compounded by the limitations of existing methods in accurately distinguishing between benign, uncertain, and pathogenic CNVs. Addressing this gap, we introduce CNVoyant, a machine learning-based multi-class framework designed to enhance the clinical significance classification of CNVs. Trained on a comprehensive dataset of 52,176 ClinVar entries across pathogenic, uncertain, and benign classifications, CNVoyant incorporates a broad spectrum of genomic features, including genome position, disease-gene annotations, dosage sensitivity, and conservation scores. Models to predict the clinical significance of copy number gains and losses were trained independently. Final models were selected after testing 29 machine learning architectures and 10,000 hyperparameter combinations each for deletions and duplications via fivefold cross-validation. We validate the performance of CNVoyant by leveraging a comprehensive set of 21,574 CNVs from the DECIPHER database, a highly regarded resource known for its extensive catalog of chromosomal imbalances linked to clinical outcomes. Compared to alternative approaches, CNVoyant shows marked improvements in precision-recall and ROC AUC metrics for binary pathogenic classifications while going one step further, offering multi-classification of clinical significance and corresponding SHAP explainability plots. Additionally, when provided germline CNV calls from real-world RGD cases with diagnostic CNV(s), CNVoyant correctly classified all diagnostic CNVs as having pathogenic significance with high confidence. This large-scale validation demonstrates CNVoyant's superior accuracy and underscores its potential to aid genomic researchers and clinical geneticists in interpreting the clinical implications of real CNVs.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


Update of
-
CNVoyant: A Highly Performant and Explainable Multi-Classifier Machine Learning Approach for Determining the Clinical Significance of Copy Number Variants.Res Sq [Preprint]. 2024 Apr 30:rs.3.rs-4308324. doi: 10.21203/rs.3.rs-4308324/v1. Res Sq. 2024. Update in: Sci Rep. 2024 Sep 28;14(1):22411. doi: 10.1038/s41598-024-72470-4. PMID: 38746157 Free PMC article. Updated. Preprint.
References
-
- Koch, L. Exploring human genomic diversity with gnomAD. Nat. Rev. Genet.21, 448–448 (2020). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
