

Uridylation regulates mRNA decay directionality in fission yeast
- PMID: 39333464
- PMCID: PMC11436920
- DOI: 10.1038/s41467-024-50824-w
Uridylation regulates mRNA decay directionality in fission yeast
Abstract
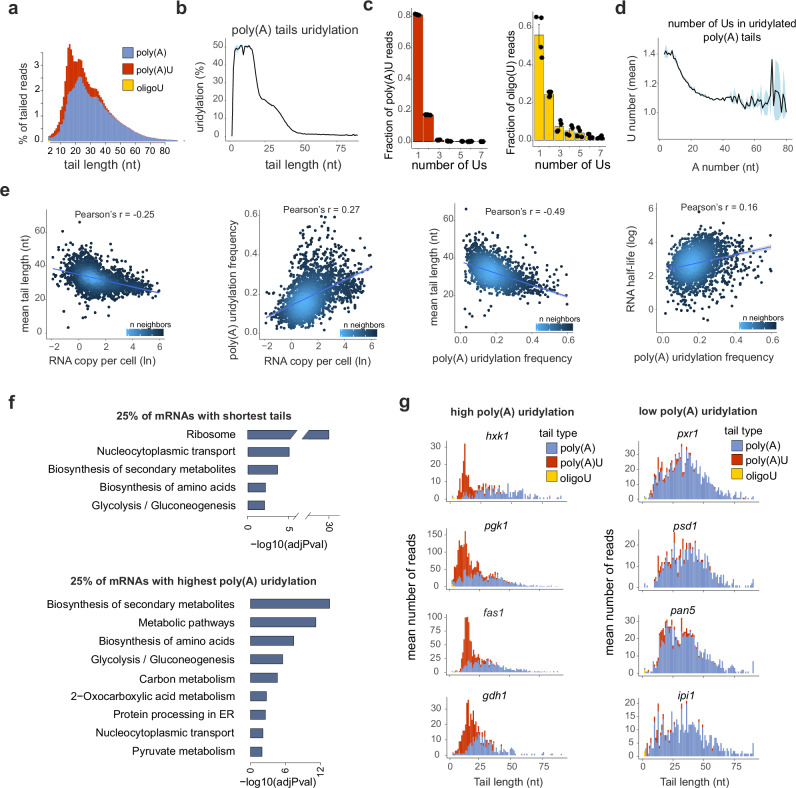
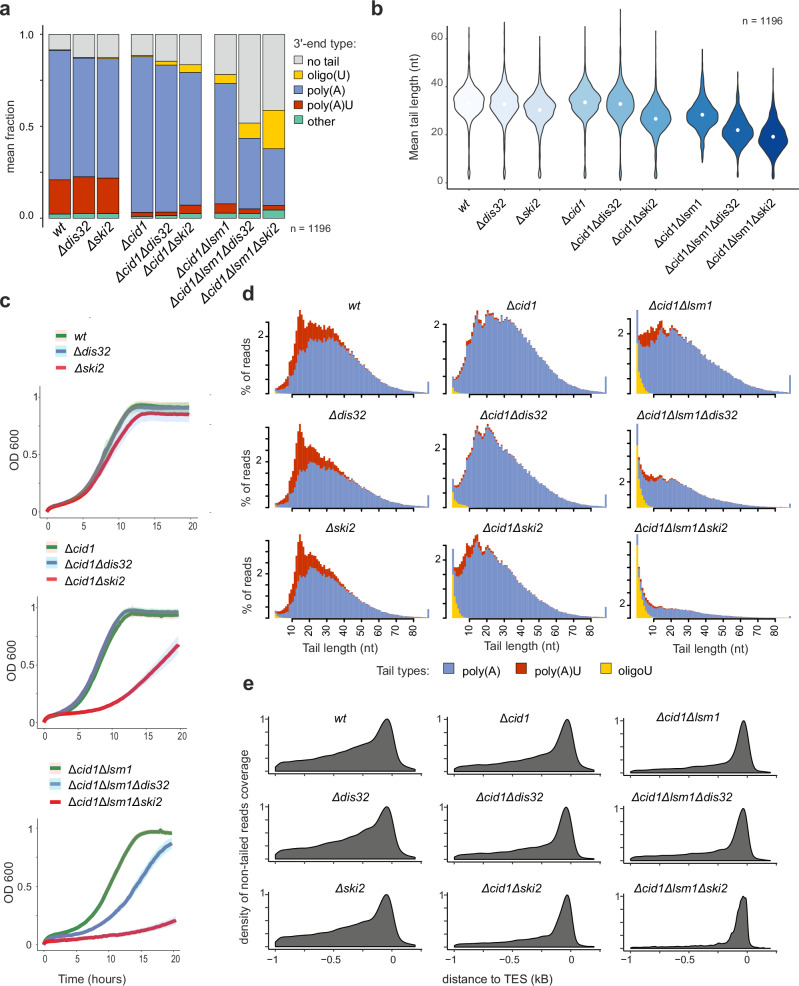
Cytoplasmic mRNA decay is effected by exonucleolytic degradation in either the 5' to 3' or 3' to 5' direction. Pervasive terminal uridylation is implicated in mRNA degradation, however, its functional relevance for bulk mRNA turnover remains poorly understood. In this study, we employ genome-wide 3'-RACE (gw3'-RACE) in the model system fission yeast to elucidate the role of uridylation in mRNA turnover. We observe widespread uridylation of shortened poly(A) tails, promoting efficient 5' to 3' mRNA decay and ensuring timely and controlled mRNA degradation. Inhibition of this uridylation process leads to excessive deadenylation and enhanced 3' to 5' mRNA decay accompanied by oligouridylation. Strikingly we found that uridylation of poly(A) tails and oligouridylation of non-polyadenylated substrates are catalysed by different terminal uridyltransferases Cid1 and Cid16 respectively. Our study sheds new light on the intricate regulatory mechanisms underlying bulk mRNA turnover, demonstrating the role of uridylation in modulating mRNA decay pathways.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- Halbach, F., Reichelt, P., Rode, M. & Conti, E. The yeast ski complex: Crystal structure and rna channeling to the exosome complex. Cell154, 814–826 (2013). - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
