

Decoding Missense Variants by Incorporating Phase Separation via Machine Learning
- PMID: 39333476
- PMCID: PMC11436885
- DOI: 10.1038/s41467-024-52580-3
Decoding Missense Variants by Incorporating Phase Separation via Machine Learning
Abstract
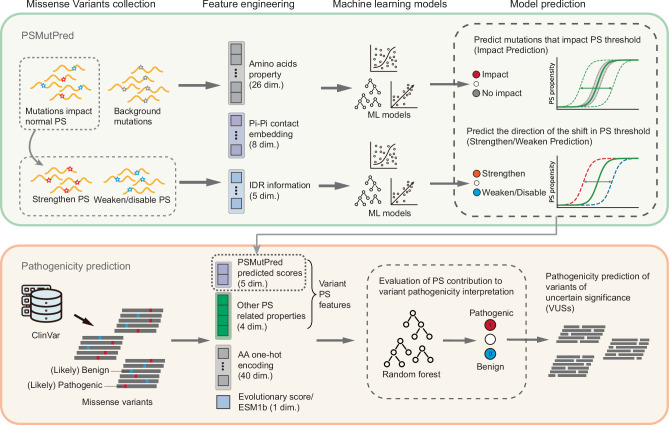
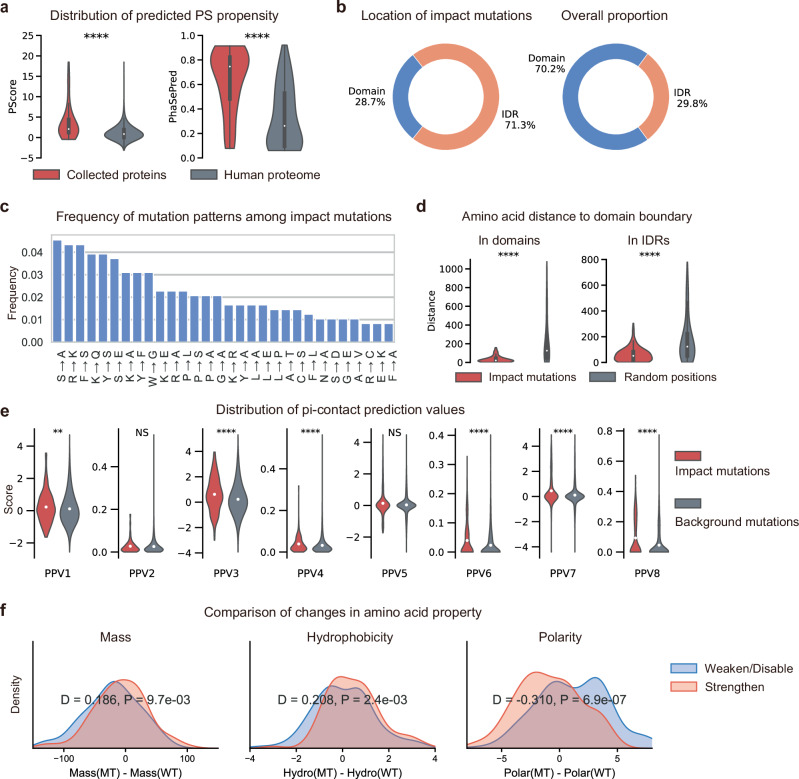
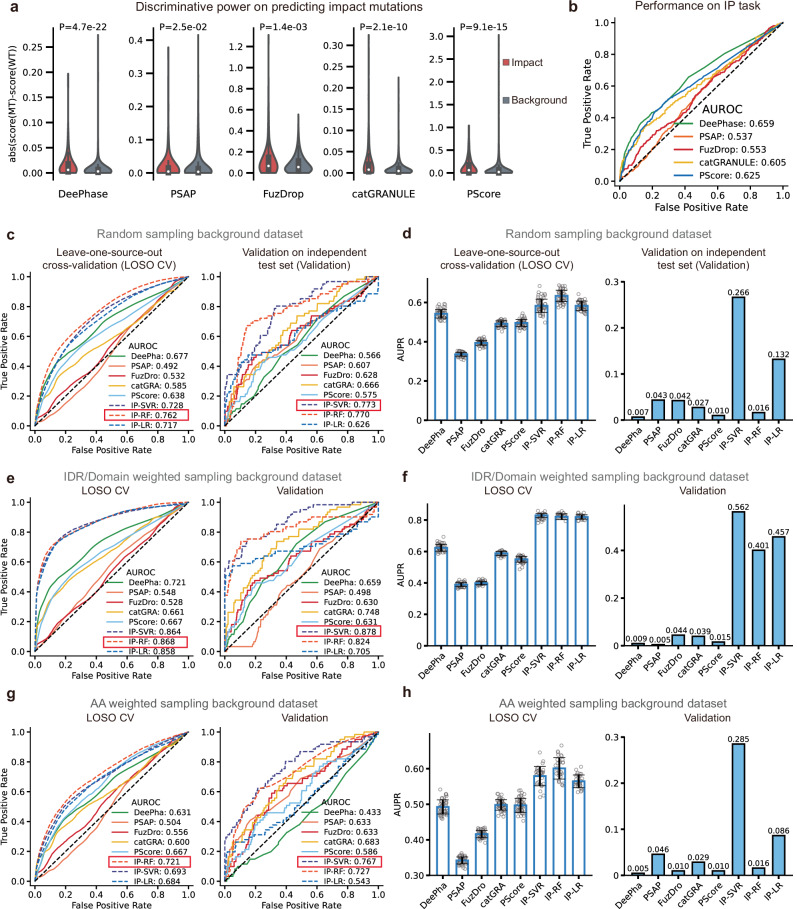
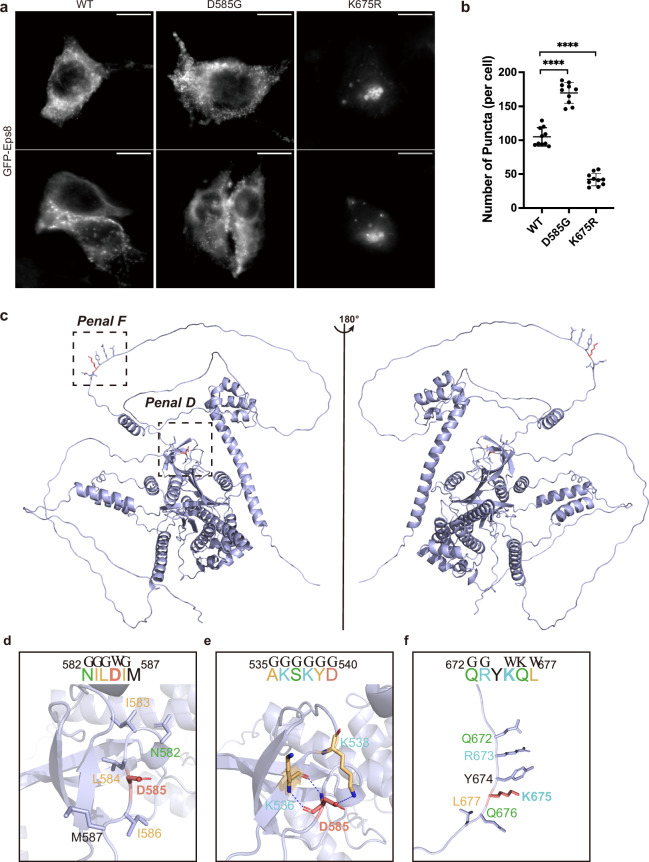
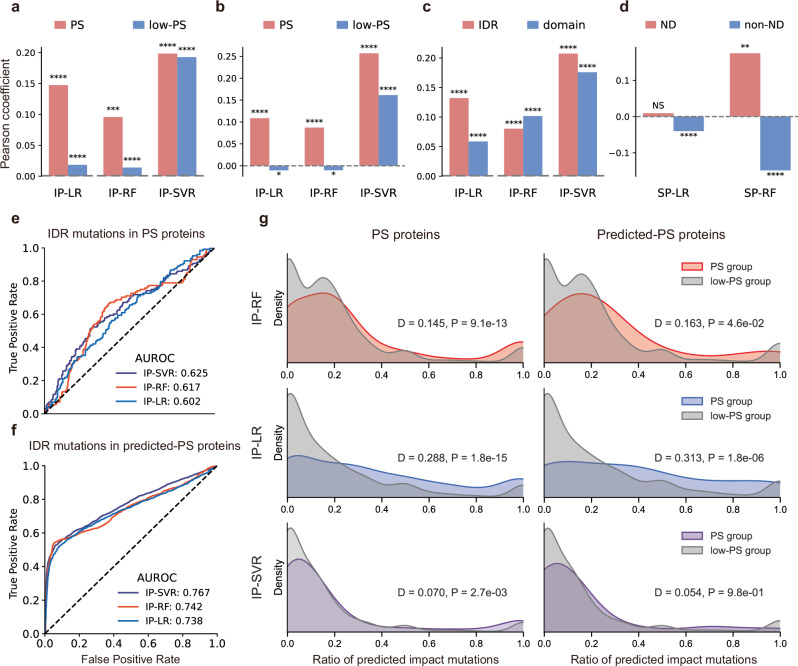
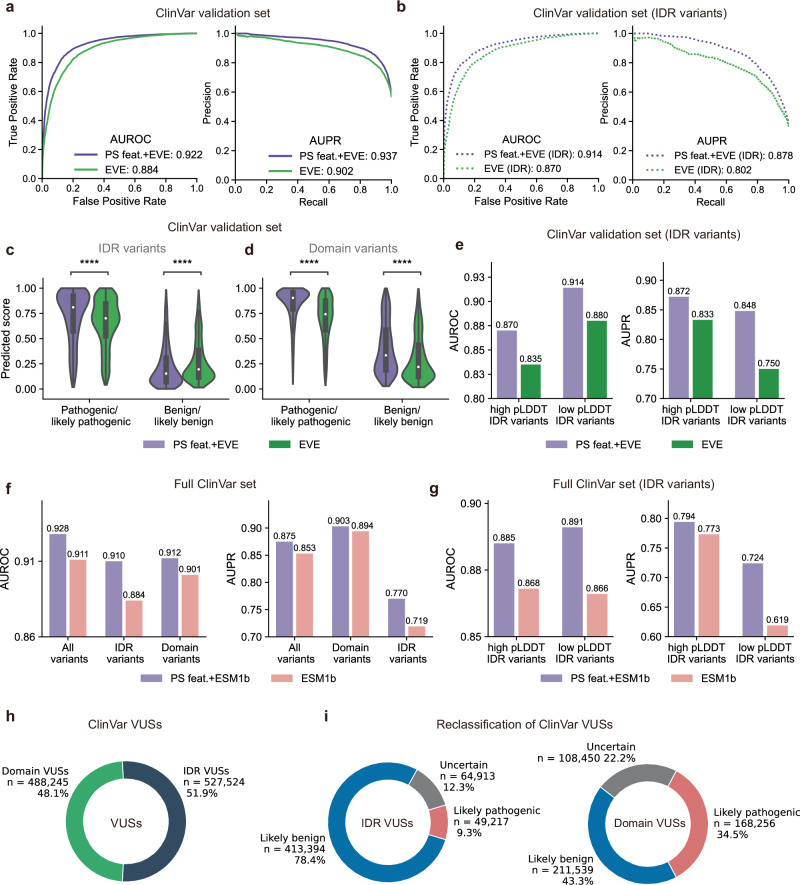
Computational models have made significant progress in predicting the effect of protein variants. However, deciphering numerous variants of uncertain significance (VUS) located within intrinsically disordered regions (IDRs) remains challenging. To address this issue, we introduce phase separation, which is tightly linked to IDRs, into the investigation of missense variants. Phase separation is vital for multiple physiological processes. By leveraging missense variants that alter phase separation propensity, we develop a machine learning approach named PSMutPred to predict the impact of missense mutations on phase separation. PSMutPred demonstrates robust performance in predicting missense variants that affect natural phase separation. In vitro experiments further underscore its validity. By applying PSMutPred on over 522,000 ClinVar missense variants, it significantly contributes to decoding the pathogenesis of disease variants, especially those in IDRs. Our work provides insights into the understanding of a vast number of VUSs in IDRs, expediting clinical interpretation and diagnosis.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
