

Towards geospatially-resolved public-health surveillance via wastewater sequencing
- PMID: 39333485
- PMCID: PMC11436780
- DOI: 10.1038/s41467-024-52427-x
Towards geospatially-resolved public-health surveillance via wastewater sequencing
Abstract
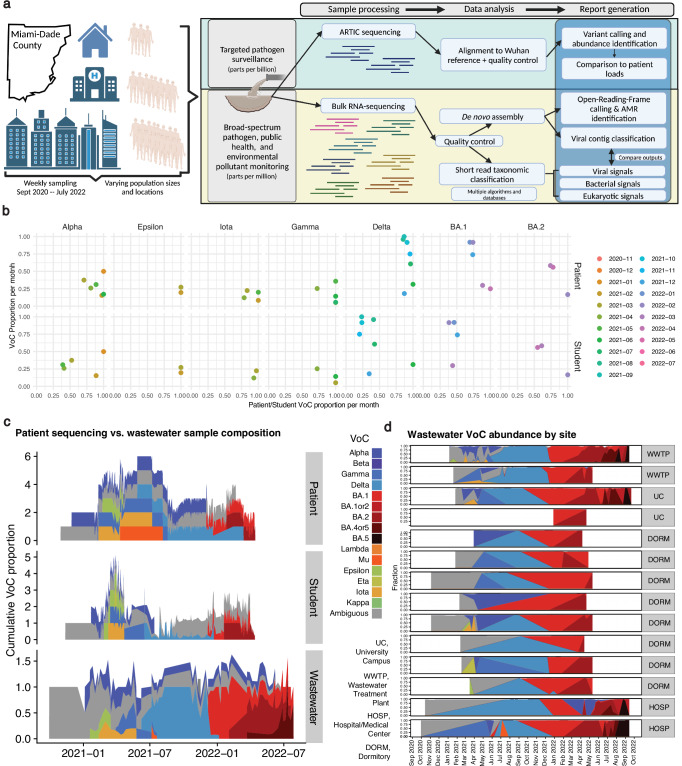
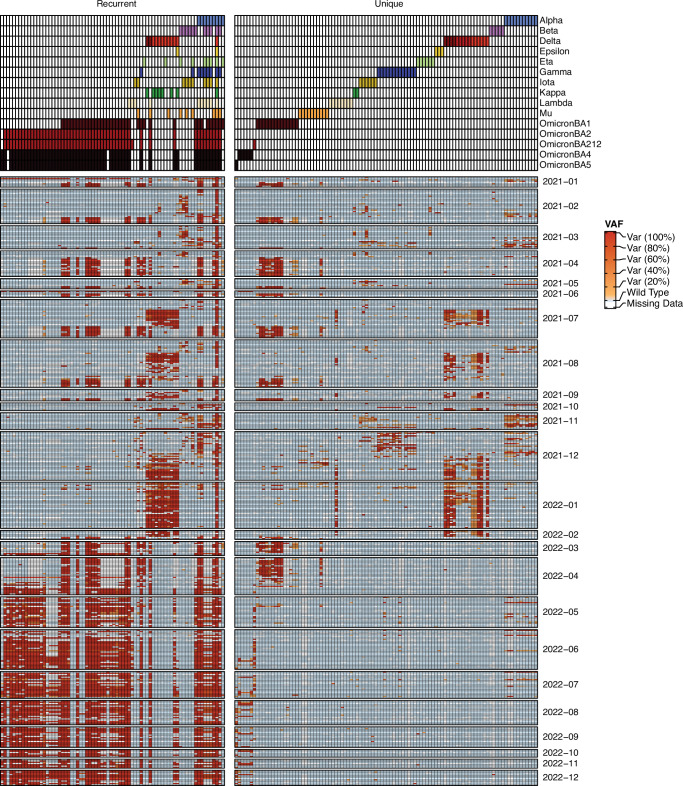
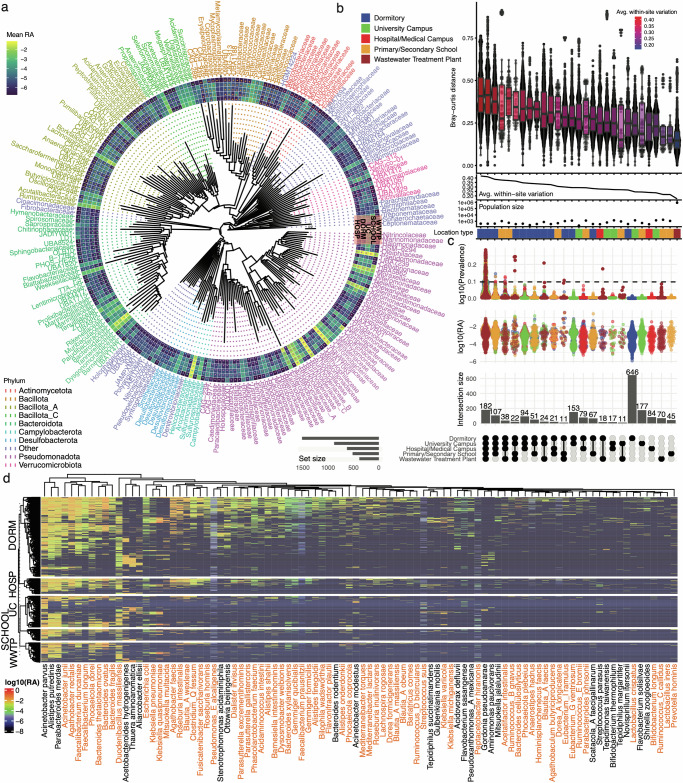
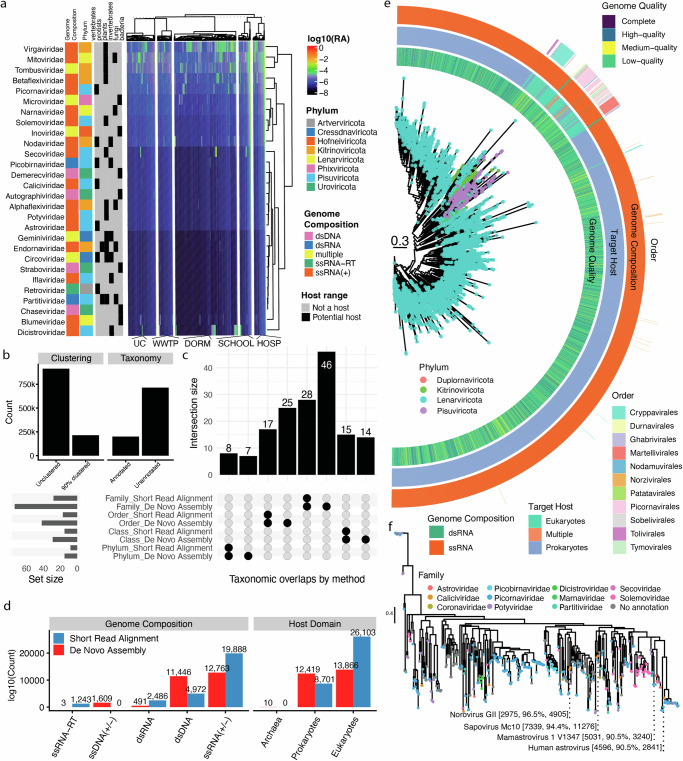
Wastewater is a geospatially- and temporally-linked microbial fingerprint of a given population, making it a potentially valuable tool for tracking public health across locales and time. Here, we integrate targeted and bulk RNA sequencing (N = 2238 samples) to track the viral, bacterial, and functional content over geospatially distinct areas within Miami Dade County, USA, from 2020-2022. We used targeted amplicon sequencing to track diverse SARS-CoV-2 variants across space and time, and we found a tight correspondence with positive PCR tests from University students and Miami-Dade hospital patients. Additionally, in bulk metatranscriptomic data, we demonstrate that the bacterial content of different wastewater sampling locations serving small population sizes can be used to detect putative, host-derived microorganisms that themselves have known associations with human health and diet. We also detect multiple enteric pathogens (e.g., Norovirus) and characterize viral diversity across sites. Moreover, we observed an enrichment of antimicrobial resistance genes (ARGs) in hospital wastewater; antibiotic-specific ARGs correlated to total prescriptions of those same antibiotics (e.g Ampicillin, Gentamicin). Overall, this effort lays the groundwork for systematic characterization of wastewater that can potentially influence public health decision-making.
© 2024. The Author(s).
Conflict of interest statement
B.T.T. is compensated for consulting with Seed Health on microbiome study design. C.E.M. is a co-founder of Onegevity and Biotia. No entity listed here was involved in funding or advising the contents of this study. G.M.C. lists competing interests at arep.med.harvard.edu/tech.html. The remaining authors declare no competing interests.
Figures
Update of
-
Geospatially-resolved public-health surveillance via wastewater sequencing.medRxiv [Preprint]. 2023 Jun 1:2023.05.31.23290781. doi: 10.1101/2023.05.31.23290781. medRxiv. 2023. Update in: Nat Commun. 2024 Sep 27;15(1):8386. doi: 10.1038/s41467-024-52427-x. PMID: 37398062 Free PMC article. Updated. Preprint.
References
-
- Ejeian, F. et al. Biosensors for wastewater monitoring: a review. Biosens. Bioelectron.118, 66–79 (2018). - PubMed
-
- O’Brien, J. W. et al. A National Wastewater Monitoring Program for a better understanding of public health: a case study using the Australian Census. Environ. Int.122, 400–411 (2019). - PubMed
-
- Leong, L. Y., Rigby, M. & Sakaji, R. H. Evaluation of the California wastewater reclamation criteria using enteric virus monitoring. Data26, 7–8 (1992).
-
- Akpor, O. B. & Muchie, B. Environmental and public health implications of wastewater quality. Afr. J. Biotechnol.10, 2379–2387 (2011).
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
