

Loss of embryonically-derived Kupffer cells during hypercholesterolemia accelerates atherosclerosis development
- PMID: 39333539
- PMCID: PMC11436809
- DOI: 10.1038/s41467-024-52735-2
Loss of embryonically-derived Kupffer cells during hypercholesterolemia accelerates atherosclerosis development
Abstract
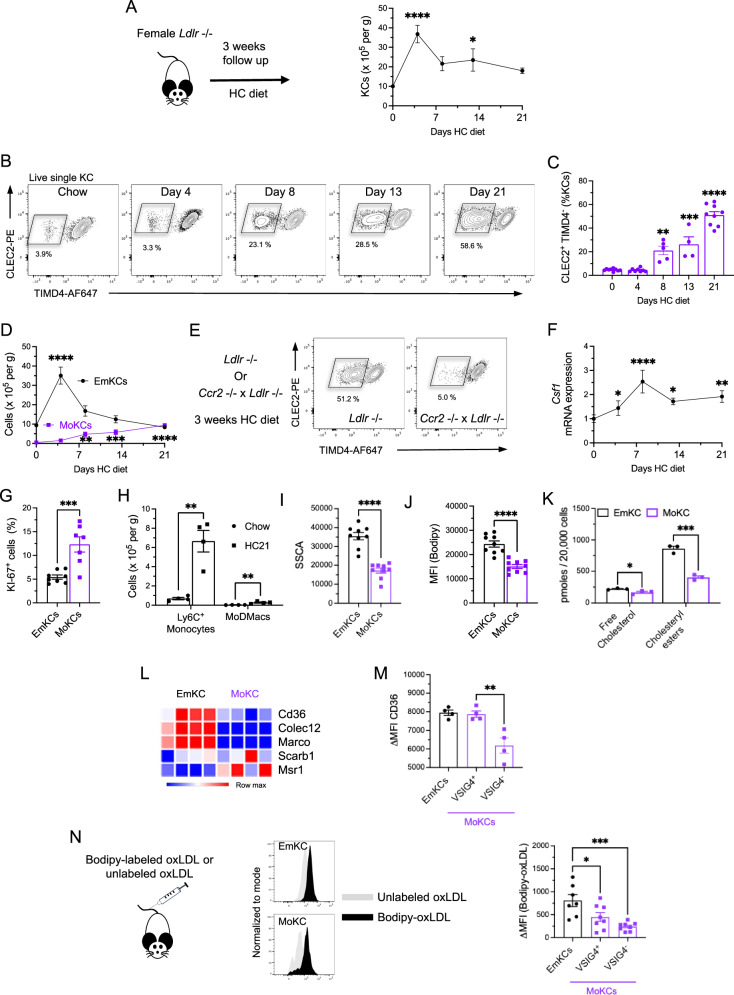
Hypercholesterolemia is a major risk factor for atherosclerosis and associated cardiovascular diseases. The liver plays a key role in the regulation of plasma cholesterol levels and hosts a large population of tissue-resident macrophages known as Kupffer cells (KCs). KCs are located in the hepatic sinusoids where they ensure key functions including blood immune surveillance. However, how KCs homeostasis is affected by the build-up of cholesterol-rich lipoproteins that occurs in the circulation during hypercholesterolemia remains unknown. Here, we show that embryo-derived KCs (EmKCs) accumulate large amounts of lipoprotein-derived cholesterol, in part through the scavenger receptor CD36, and massively expand early after the induction of hypercholesterolemia. After this rapid adaptive response, EmKCs exhibit mitochondrial oxidative stress and their numbers gradually diminish while monocyte-derived KCs (MoKCs) with reduced cholesterol-loading capacities seed the KC pool. Decreased proportion of EmKCs in the KC pool enhances liver cholesterol content and exacerbates hypercholesterolemia, leading to accelerated atherosclerotic plaque development. Together, our data reveal that KC homeostasis is perturbed during hypercholesterolemia, which in turn alters the control of plasma cholesterol levels and increases atherosclerosis.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
