

Proteome-wide copy-number estimation from transcriptomics
- PMID: 39333715
- PMCID: PMC11535397
- DOI: 10.1038/s44320-024-00064-3
Proteome-wide copy-number estimation from transcriptomics
Abstract
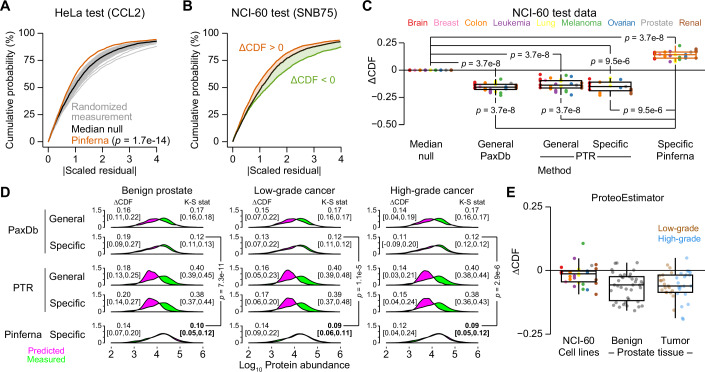
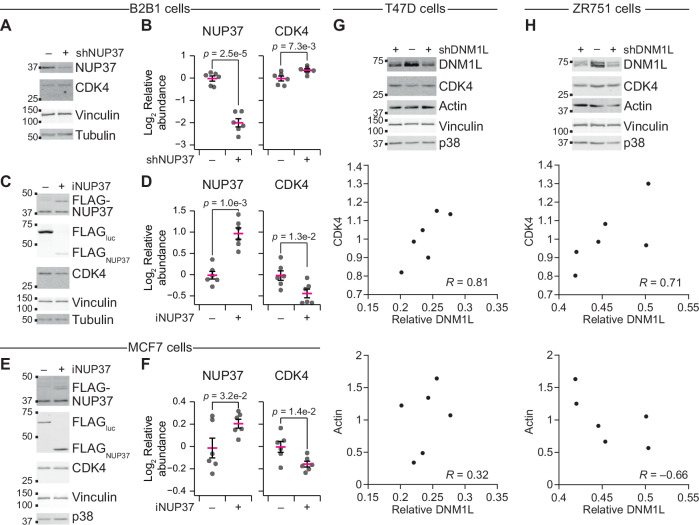
Protein copy numbers constrain systems-level properties of regulatory networks, but proportional proteomic data remain scarce compared to RNA-seq. We related mRNA to protein statistically using best-available data from quantitative proteomics and transcriptomics for 4366 genes in 369 cell lines. The approach starts with a protein's median copy number and hierarchically appends mRNA-protein and mRNA-mRNA dependencies to define an optimal gene-specific model linking mRNAs to protein. For dozens of cell lines and primary samples, these protein inferences from mRNA outmatch stringent null models, a count-based protein-abundance repository, empirical mRNA-to-protein ratios, and a proteogenomic DREAM challenge winner. The optimal mRNA-to-protein relationships capture biological processes along with hundreds of known protein-protein complexes, suggesting mechanistic relationships. We use the method to identify a viral-receptor abundance threshold for coxsackievirus B3 susceptibility from 1489 systems-biology infection models parameterized by protein inference. When applied to 796 RNA-seq profiles of breast cancer, inferred copy-number estimates collectively re-classify 26-29% of luminal tumors. By adopting a gene-centered perspective of mRNA-protein covariation across different biological contexts, we achieve accuracies comparable to the technical reproducibility of contemporary proteomics.
Keywords: CCLE; CVB3; Pinferna; SWATH; TMT.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures








Update of
-
Proteome-wide copy-number estimation from transcriptomics.bioRxiv [Preprint]. 2023 Jul 11:2023.07.10.548432. doi: 10.1101/2023.07.10.548432. bioRxiv. 2023. Update in: Mol Syst Biol. 2024 Nov;20(11):1230-1256. doi: 10.1038/s44320-024-00064-3. PMID: 37503057 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
