

DNA damage response in breast cancer and its significant role in guiding novel precise therapies
- PMID: 39334297
- PMCID: PMC11437670
- DOI: 10.1186/s40364-024-00653-2
DNA damage response in breast cancer and its significant role in guiding novel precise therapies
Abstract
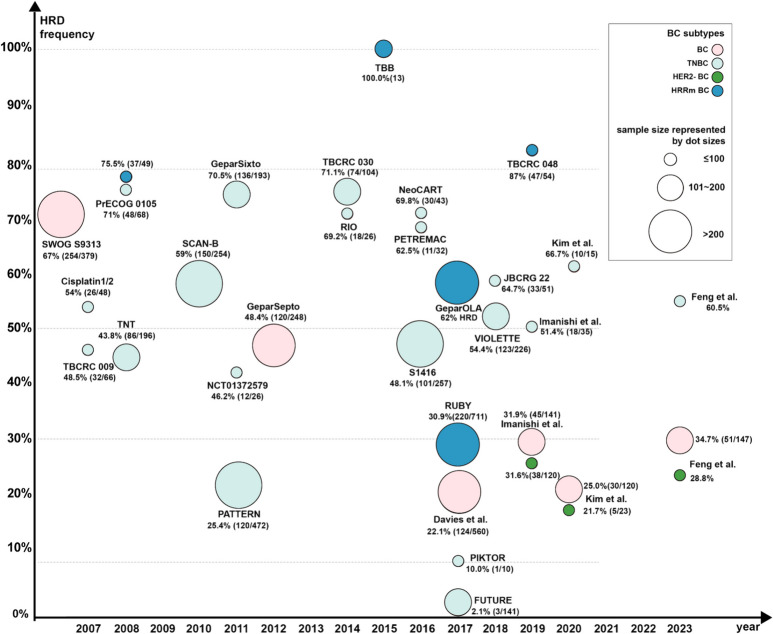
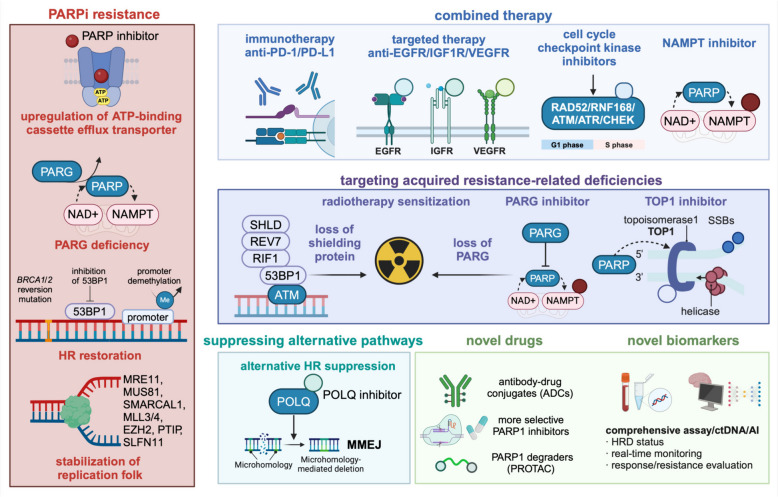
DNA damage response (DDR) deficiency has been one of the emerging targets in treating breast cancer in recent years. On the one hand, DDR coordinates cell cycle and signal transduction, whose dysfunction may lead to cell apoptosis, genomic instability, and tumor development. Conversely, DDR deficiency is an intrinsic feature of tumors that underlies their response to treatments that inflict DNA damage. In this review, we systematically explore various mechanisms of DDR, the rationale and research advances in DDR-targeted drugs in breast cancer, and discuss the challenges in its clinical applications. Notably, poly (ADP-ribose) polymerase (PARP) inhibitors have demonstrated favorable efficacy and safety in breast cancer with high homogenous recombination deficiency (HRD) status in a series of clinical trials. Moreover, several studies on novel DDR-related molecules are actively exploring to target tumors that become resistant to PARP inhibition. Before further clinical application of new regimens or drugs, novel and standardized biomarkers are needed to develop for accurately characterizing the benefit population and predicting efficacy. Despite the promising efficacy of DDR-related treatments, challenges of off-target toxicity and drug resistance need to be addressed. Strategies to overcome drug resistance await further exploration on DDR mechanisms, and combined targeted drugs or immunotherapy will hopefully provide more precise or combined strategies and expand potential responsive populations.
Keywords: Breast cancer; DNA damage response; Homogenous recombination deficiency; Poly (ADP-ribose) polymerase (PARP) inhibitor.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
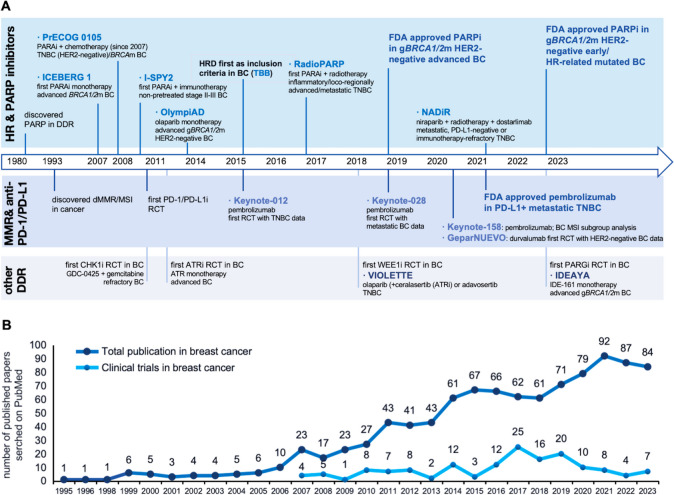
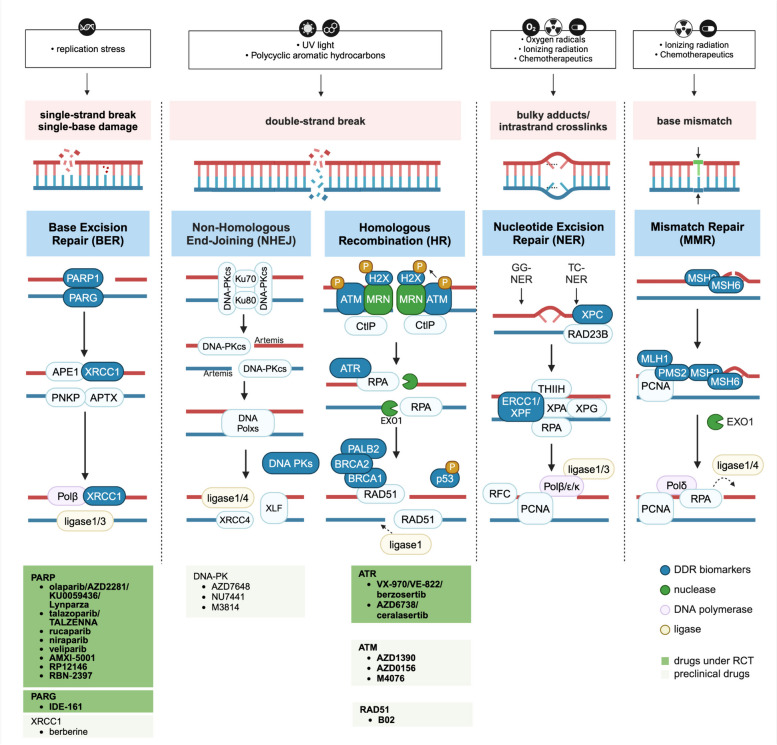
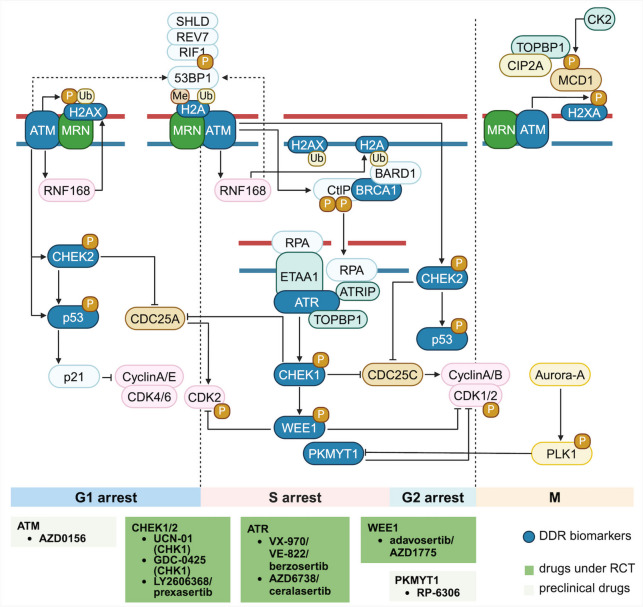
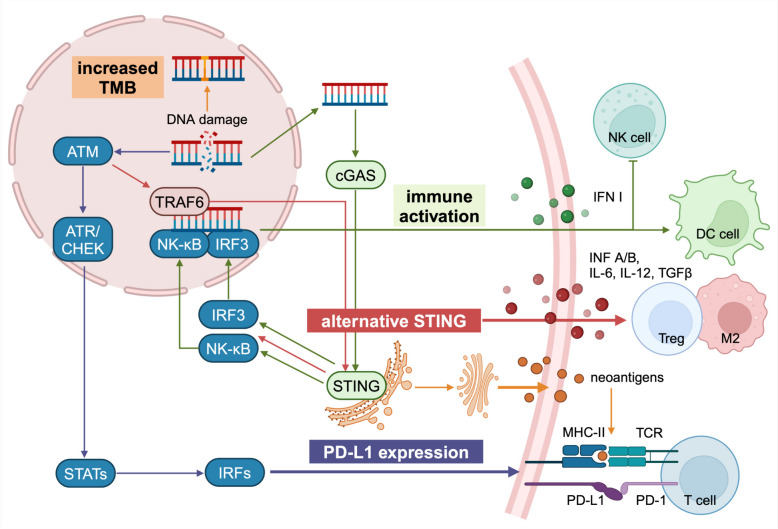
References
Publication types
LinkOut - more resources
Full Text Sources
