

Factors influencing survival in sphingosine phosphate lyase insufficiency syndrome: a retrospective cross-sectional natural history study of 76 patients
- PMID: 39334450
- PMCID: PMC11429486
- DOI: 10.1186/s13023-024-03311-w
Factors influencing survival in sphingosine phosphate lyase insufficiency syndrome: a retrospective cross-sectional natural history study of 76 patients
Abstract
Background: Sphingosine-1-phosphate lyase insufficiency syndrome (SPLIS) is a recently recognized inborn error of metabolism associated with steroid-resistant nephrotic syndrome as well as adrenal insufficiency and immunological, neurological, and skin manifestations. SPLIS is caused by inactivating mutations in SGPL1, encoding the pyridoxal 5'phosphate-dependent enzyme sphingosine-1-phosphate lyase, which catalyzes the final step of sphingolipid metabolism. Some SPLIS patients have undergone kidney transplantation, and others have been treated with vitamin B6 supplementation. In addition, targeted therapies including gene therapy are in preclinical development. In anticipation of clinical trials, it will be essential to characterize the full spectrum and natural history of SPLIS. We performed a retrospective analysis of 76 patients in whom the diagnosis of SPLIS was established in a proband with at least one suggestive finding and biallelic SGPL1 variants identified by molecular genetic testing. The main objective of the study was to identify factors influencing survival in SPLIS subjects.
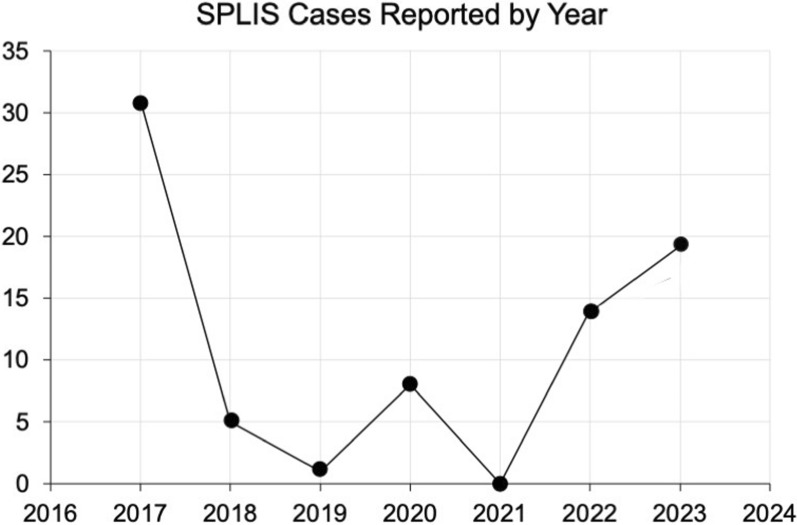
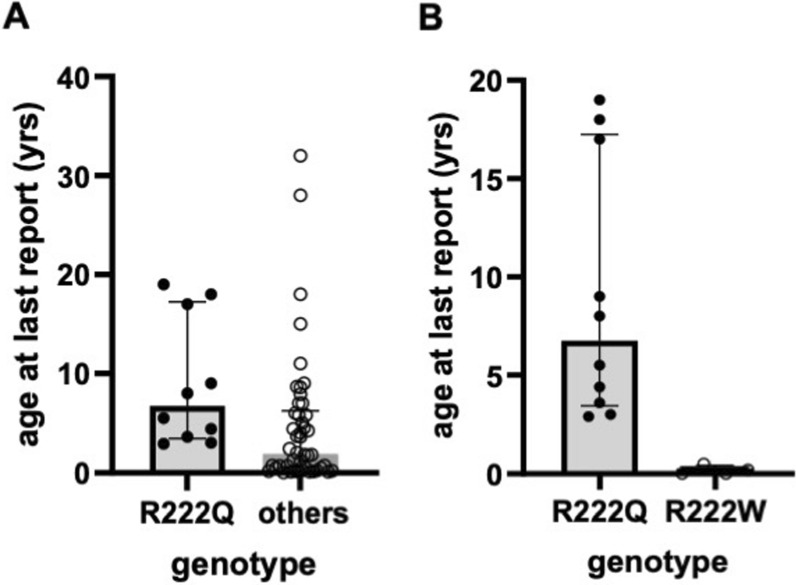
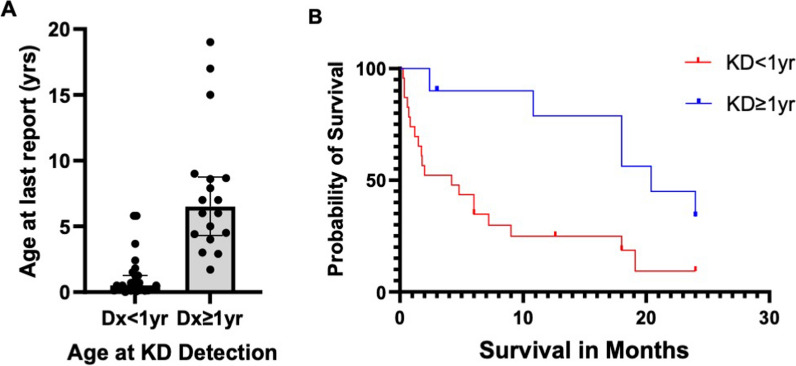
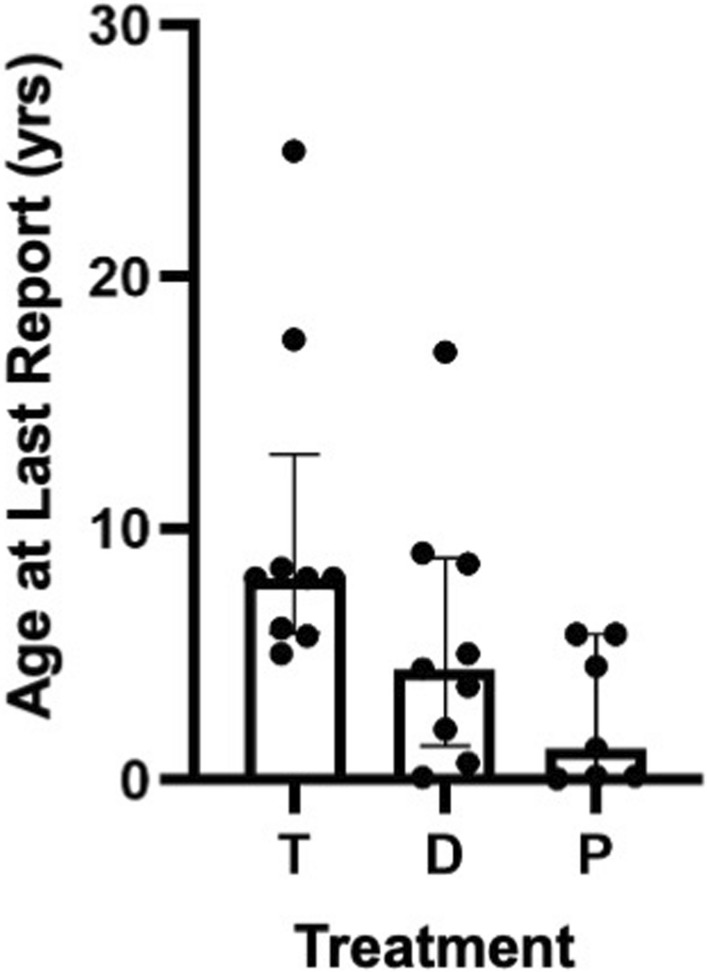
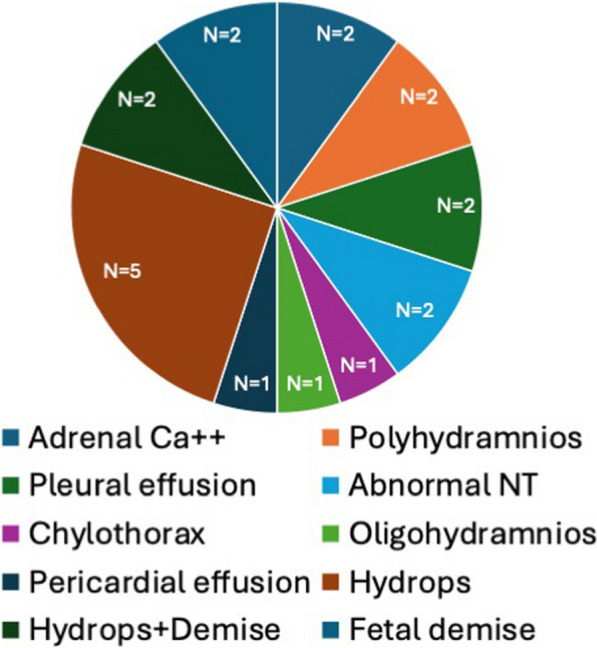
Results: Overall survival at last report was 50%. Major influences on survival included: (1) age and organ involvement at first presentation; (2) receiving a kidney transplant, and (3) SGPL1 genotype. Among 48 SPLIS patients with nephropathy who had not received a kidney transplant, two clinical subgroups were distinguished. Of children diagnosed with SPLIS nephropathy before age one (n = 30), less than 30% were alive 2 years after diagnosis, and 17% were living at last report. Among those diagnosed at or after age one (n = 18), ~ 70% were alive 2 years after diagnosis, and 72% were living at time of last report. SPLIS patients homozygous for the SPL R222Q variant survived longer compared to patients with other genotypes. Kidney transplantation significantly extended survival outcomes.
Conclusion: Our results demonstrate that SPLIS is a phenotypically heterogeneous condition. We find that patients diagnosed with SPLIS nephropathy in the first year of life and patients presenting with prenatal findings represent two high-risk subgroups, whereas patients harboring the R222Q SGPL1 variant fare better than the rest. Time to progression from onset of proteinuria to end stage kidney disease varies from less than one month to five years, and kidney transplantation may be lifesaving.
Keywords: SGPL1; Adrenal insufficiency; Gene therapy; Inborn error of metabolism; Kidney transplantation; Nephrotic syndrome; Pyridoxal 5′-phosphate; SPLIS; Vitamin B6.
© 2024. The Author(s).
Conflict of interest statement
J.D.S. is an author on patent International Application Serial No. PCT/US2021/018613, “Adeno-Associated Viral (Aav)-Mediated
Figures
References
-
- Weaver KN, Sullivan B, Hildebrandt F, et al. Sphingosine phosphate lyase insufficiency syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews. Seattle (WA), 2020. - PubMed
-
- Pournasiri Z, Madani A, Nazarpack F, et al. Sphingosine phosphate lyase insufficiency syndrome: a systematic review. World J Pediatr. 2022. 10.1007/s12519-022-00615-4. - PubMed
-
- Tastemel Ozturk T, Canpolat N, Saygili S, et al. A rare cause of nephrotic syndrome-sphingosine-1-phosphate lyase (SGPL1) deficiency: 6 cases and a review of the literature. Pediatr Nephrol. 2023;38(3):711–9. 10.1007/s00467-022-05656-5. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
