

Quintessential Synergy: Concurrent Transient Administration of Integrated Stress Response Inhibitors and BACE1 and/or BACE2 Activators as the Optimal Therapeutic Strategy for Alzheimer's Disease
- PMID: 39337400
- PMCID: PMC11432332
- DOI: 10.3390/ijms25189913
Quintessential Synergy: Concurrent Transient Administration of Integrated Stress Response Inhibitors and BACE1 and/or BACE2 Activators as the Optimal Therapeutic Strategy for Alzheimer's Disease
Abstract
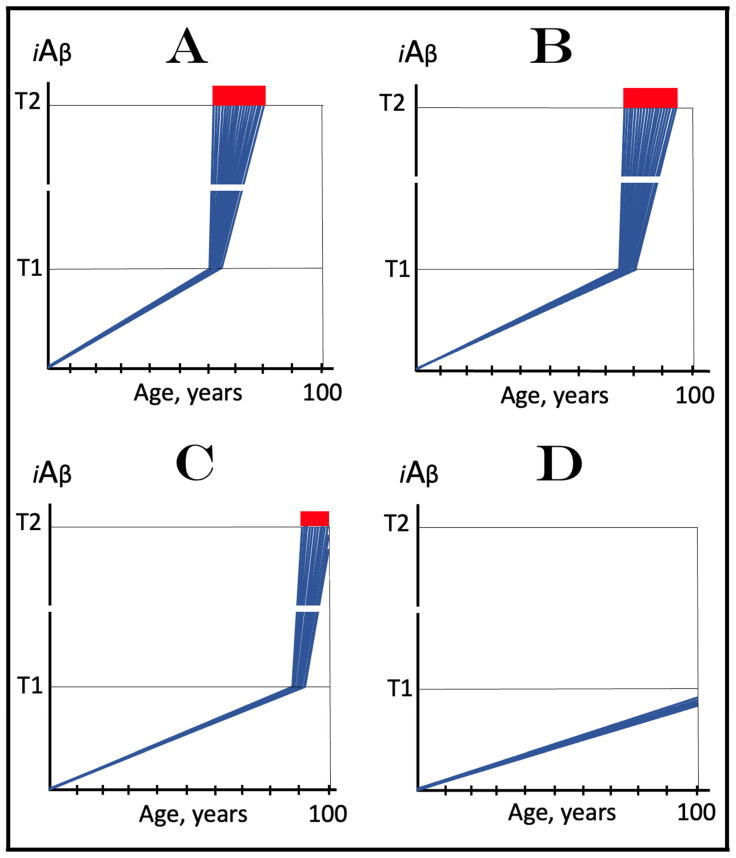
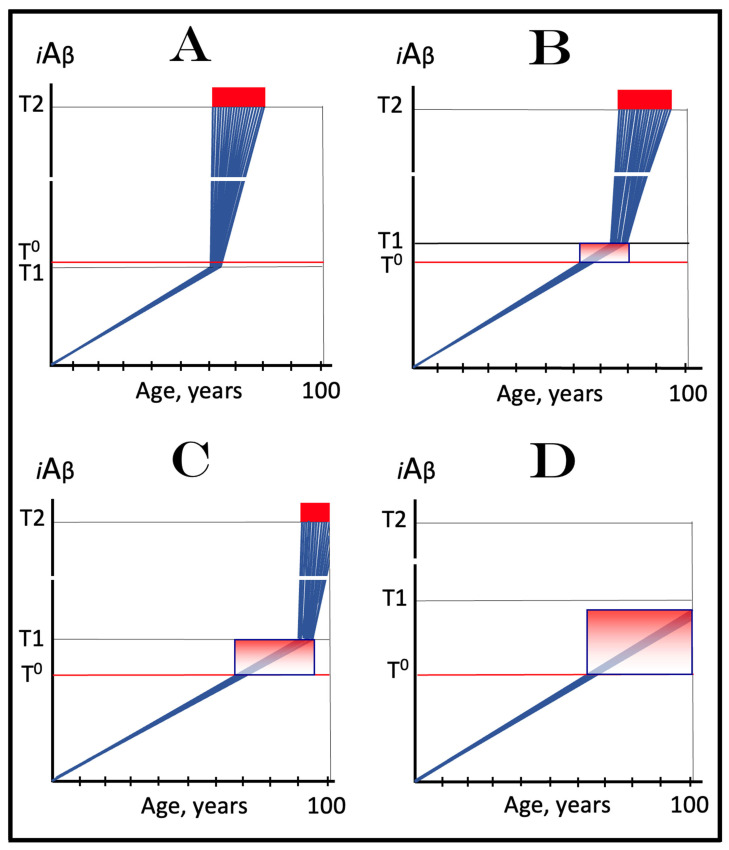
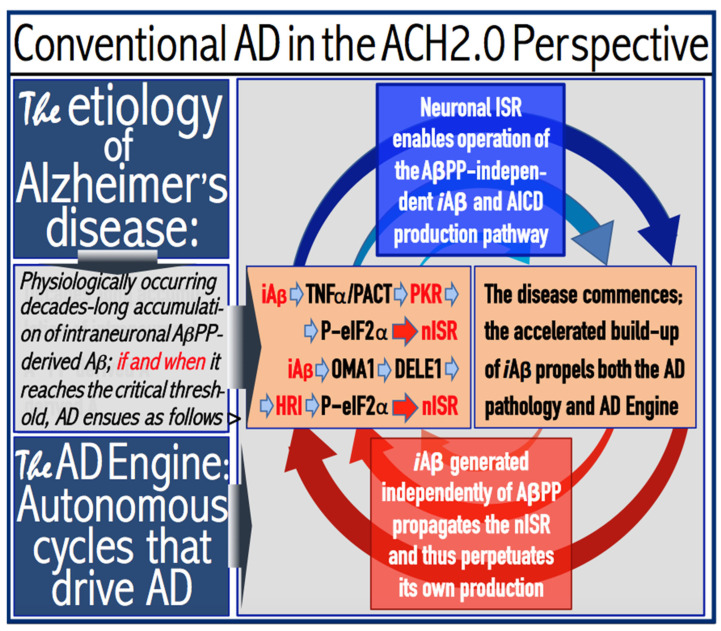
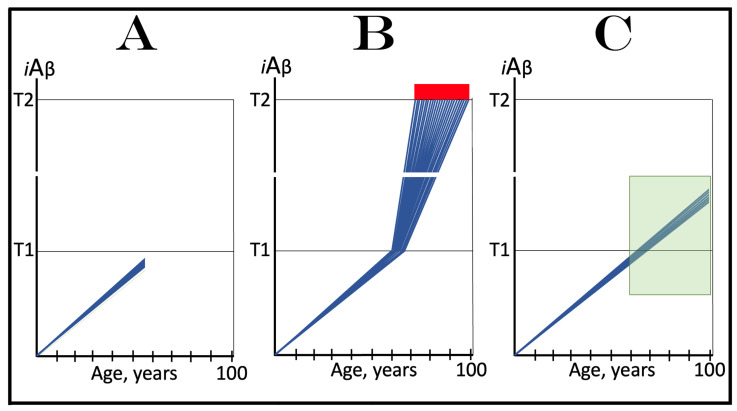
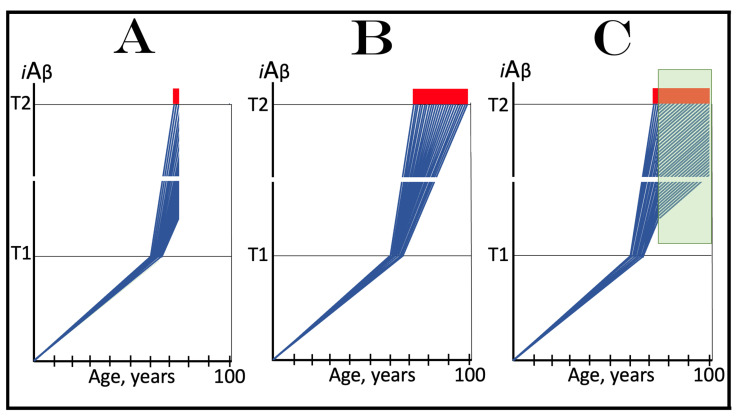
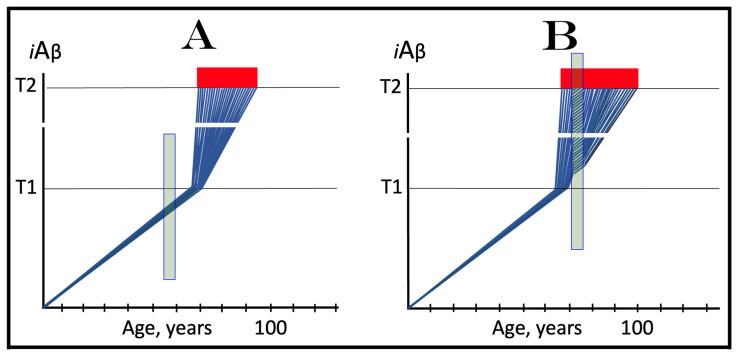
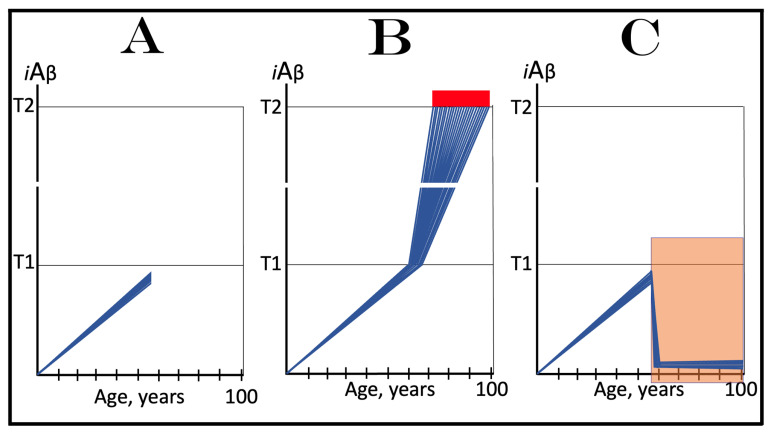
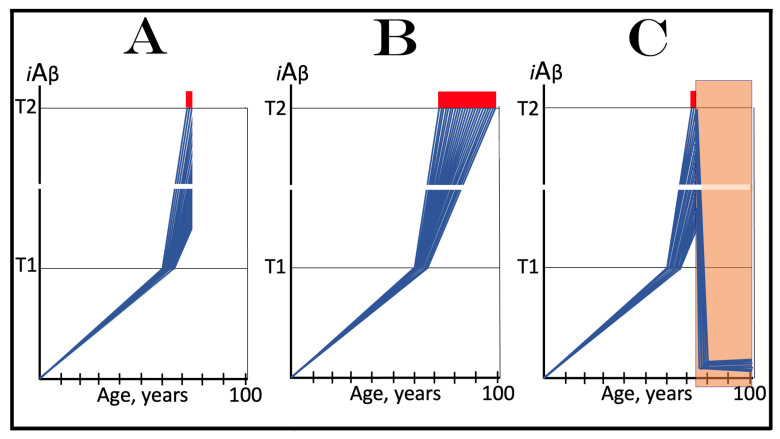
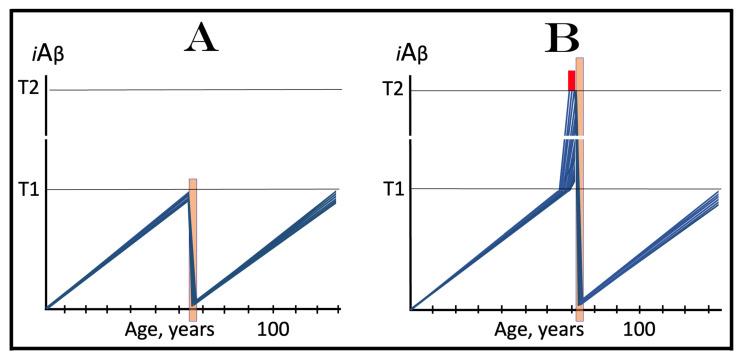
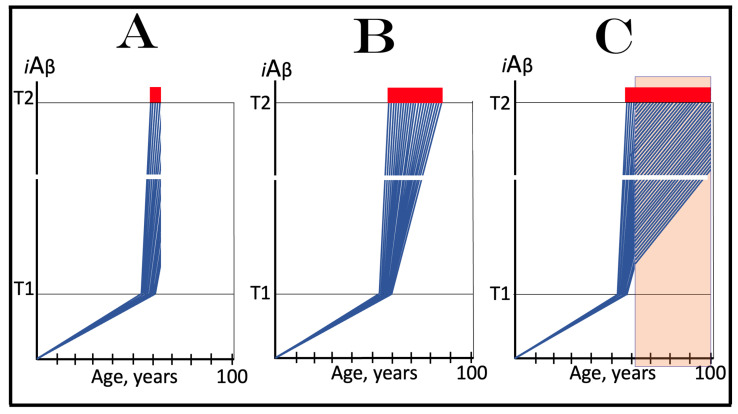
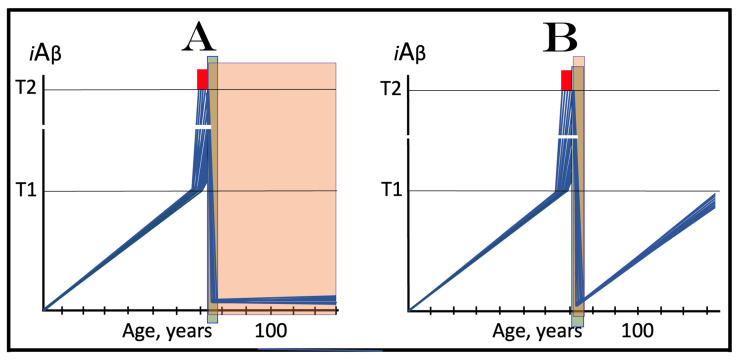
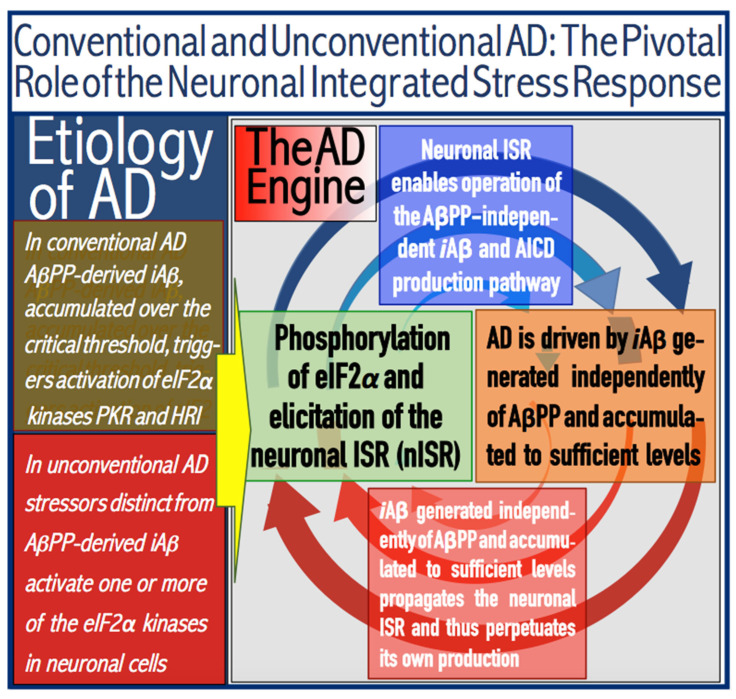
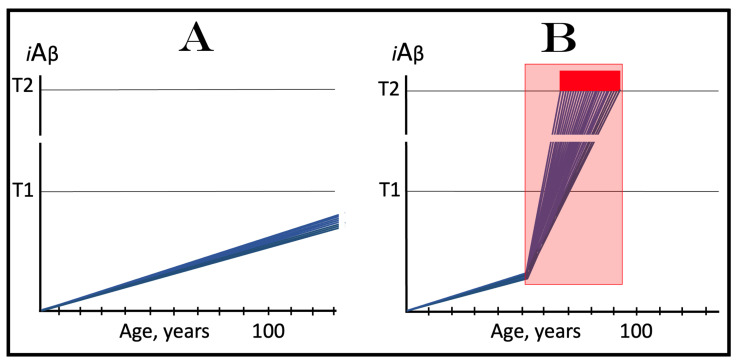
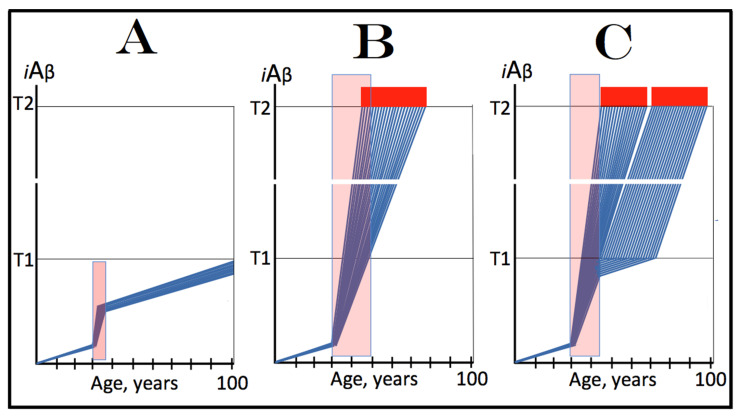
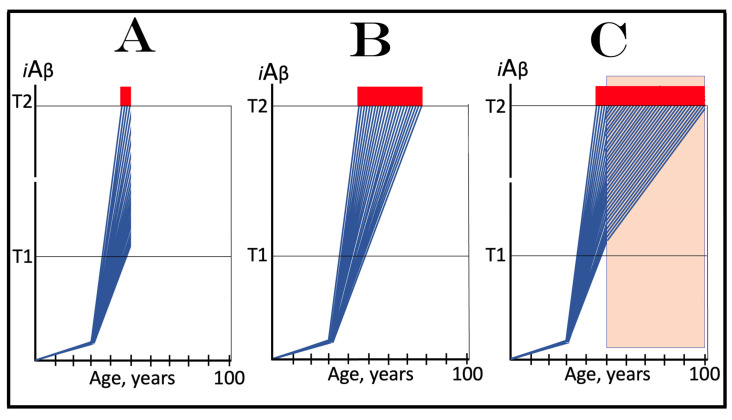
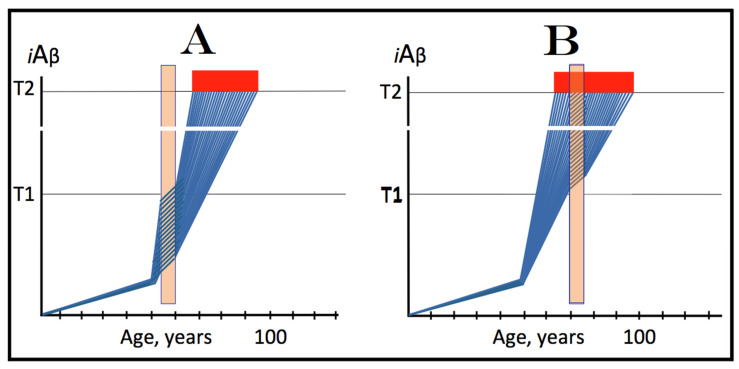
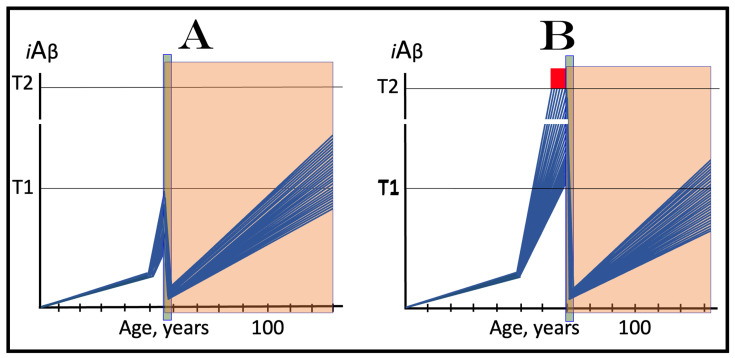
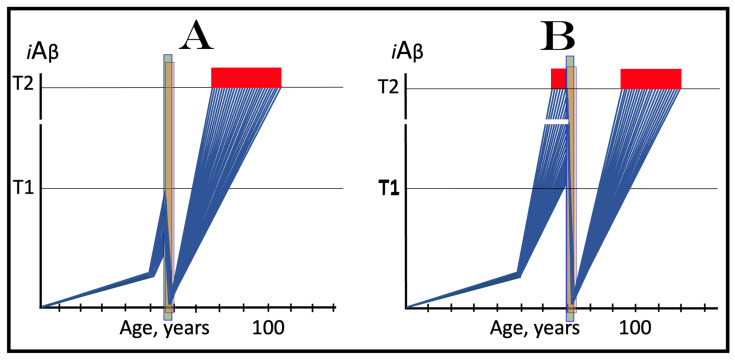
The present study analyzes two potential therapeutic approaches for Alzheimer's disease (AD). One is the suppression of the neuronal integrated stress response (ISR). Another is the targeted degradation of intraneuronal amyloid-beta (iAβ) via the activation of BACE1 (Beta-site Aβ-protein-precursor Cleaving Enzyme) and/or BACE2. Both approaches are rational. Both are promising. Both have substantial intrinsic limitations. However, when combined in a carefully orchestrated manner into a composite therapy they display a prototypical synergy and constitute the apparently optimal, potentially most effective therapeutic strategy for AD.
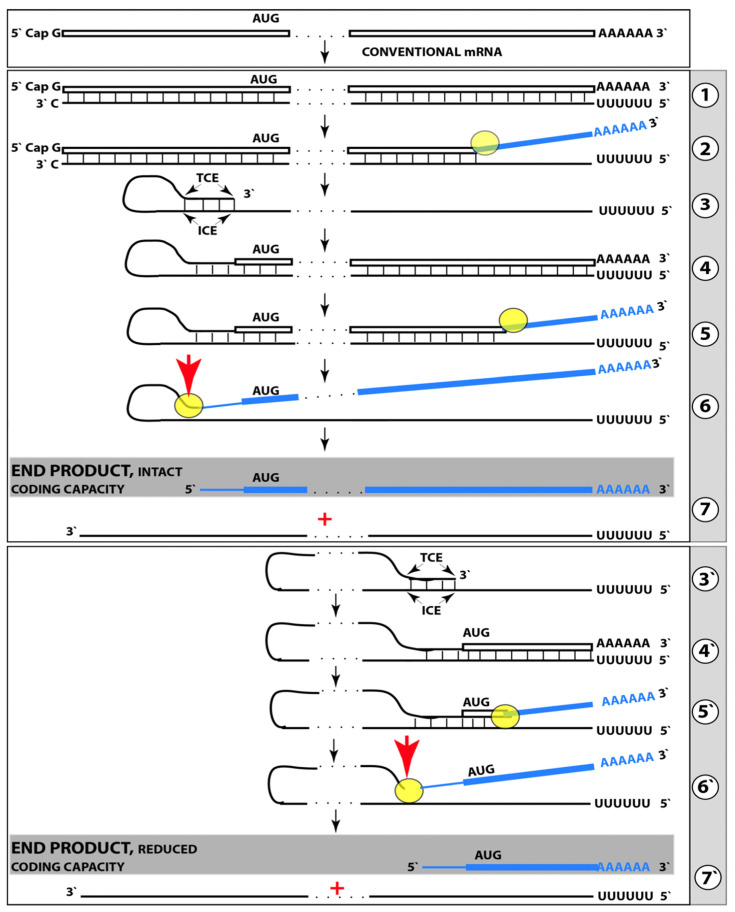
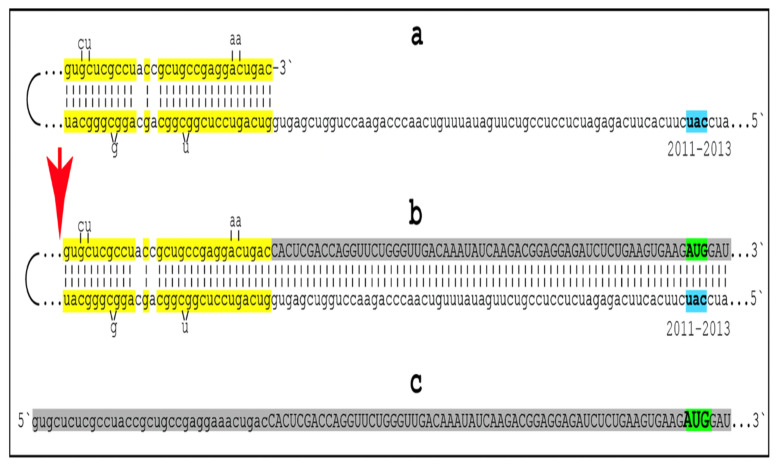
Keywords: AβPP-independent generation of iAβ; RNA-dependent asymmetric amplification of human AβPP mRNA; amyloid cascade hypothesis 2.0 (ACH2.0); conventional and unconventional Alzheimer’s disease; depletion of iAβ via the activation of BACE1 and/or BACE2; initiation of translation from the AUG codon encoding Met671 of the intact or 5′-truncated human AβPP mRNA; intraneuronal Aβ (iAβ); neuronal integrated stress response (ISR); suppression of the neuronal ISR; therapeutic strategies for conventional and unconventional Alzheimer’s disease.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
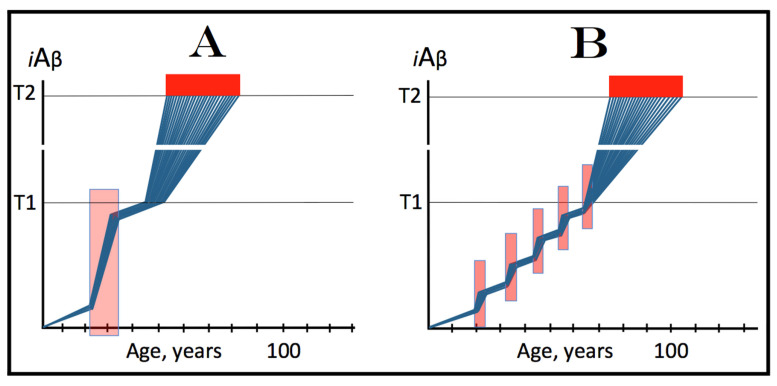
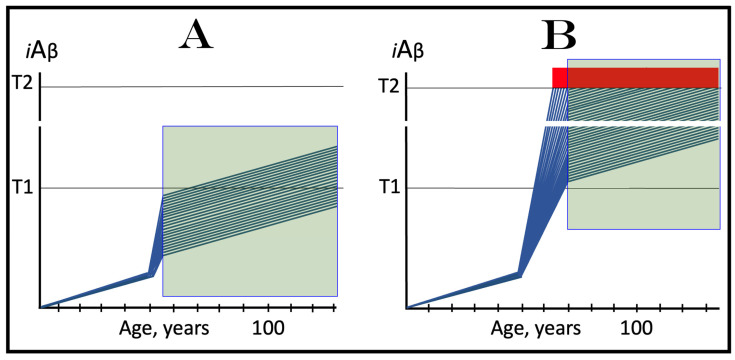
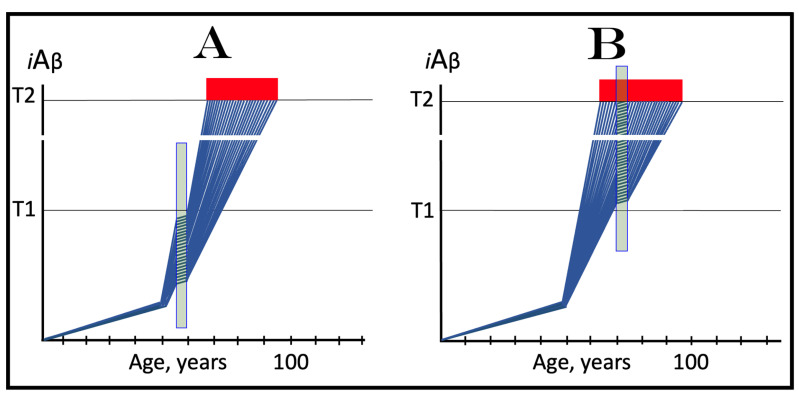
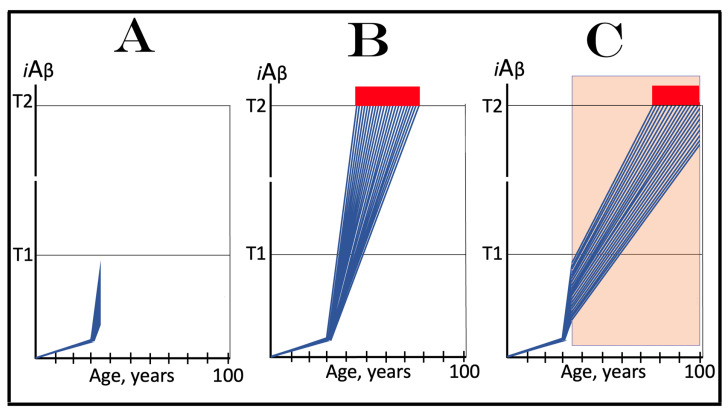
References
-
- Volloch V., Rits-Volloch S. Principles of Design of Clinical Trials for Prevention and Treatment of Alzheimer’s Disease and Aging-Associated Cognitive Decline in the ACH2.0 Perspective: Potential Outcomes, Challenges and Solutions. J. Alzheimer’s Dis. Rep. 2023;7:921–955. doi: 10.3233/ADR-230037. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
