

Combining Nanopore direct RNA sequencing with genetics and mass spectrometry for analysis of T-loop base modifications across 42 yeast tRNA isoacceptors
- PMID: 39340295
- PMCID: PMC11514469
- DOI: 10.1093/nar/gkae796
Combining Nanopore direct RNA sequencing with genetics and mass spectrometry for analysis of T-loop base modifications across 42 yeast tRNA isoacceptors
Abstract
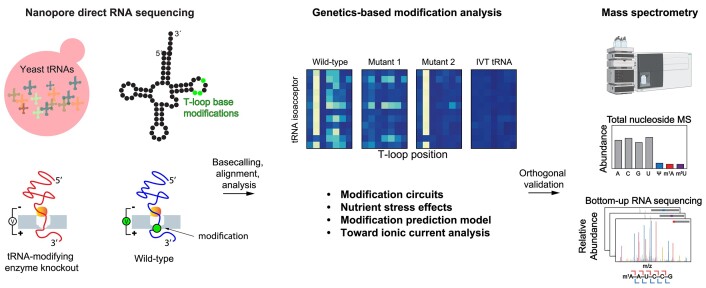
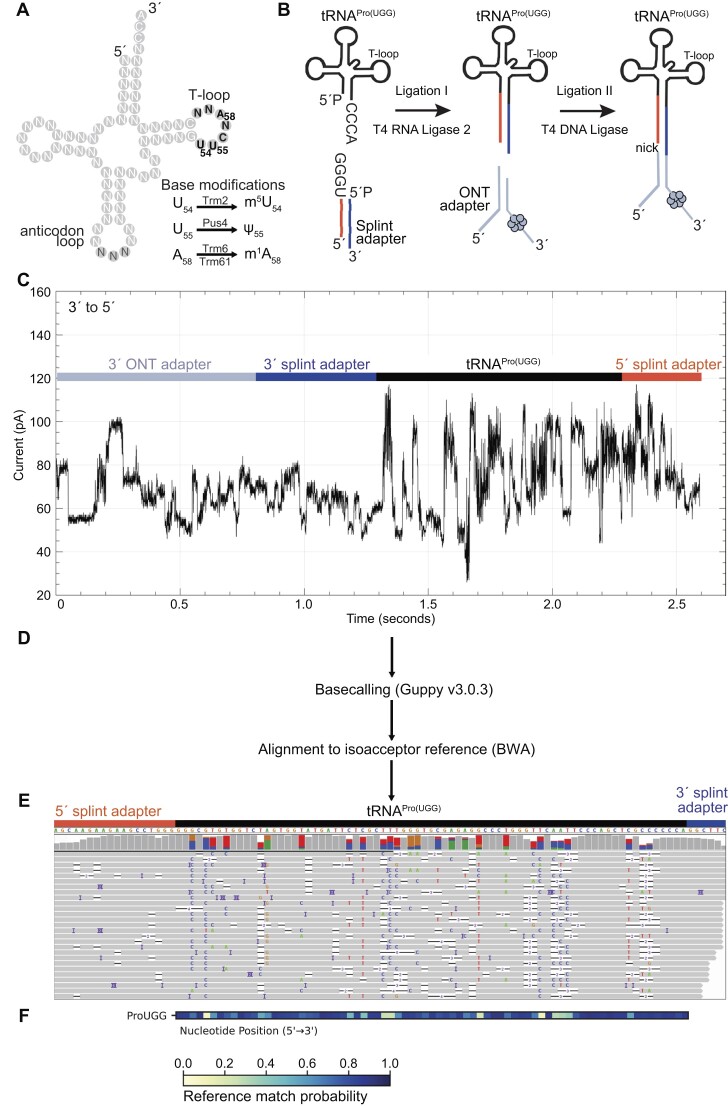
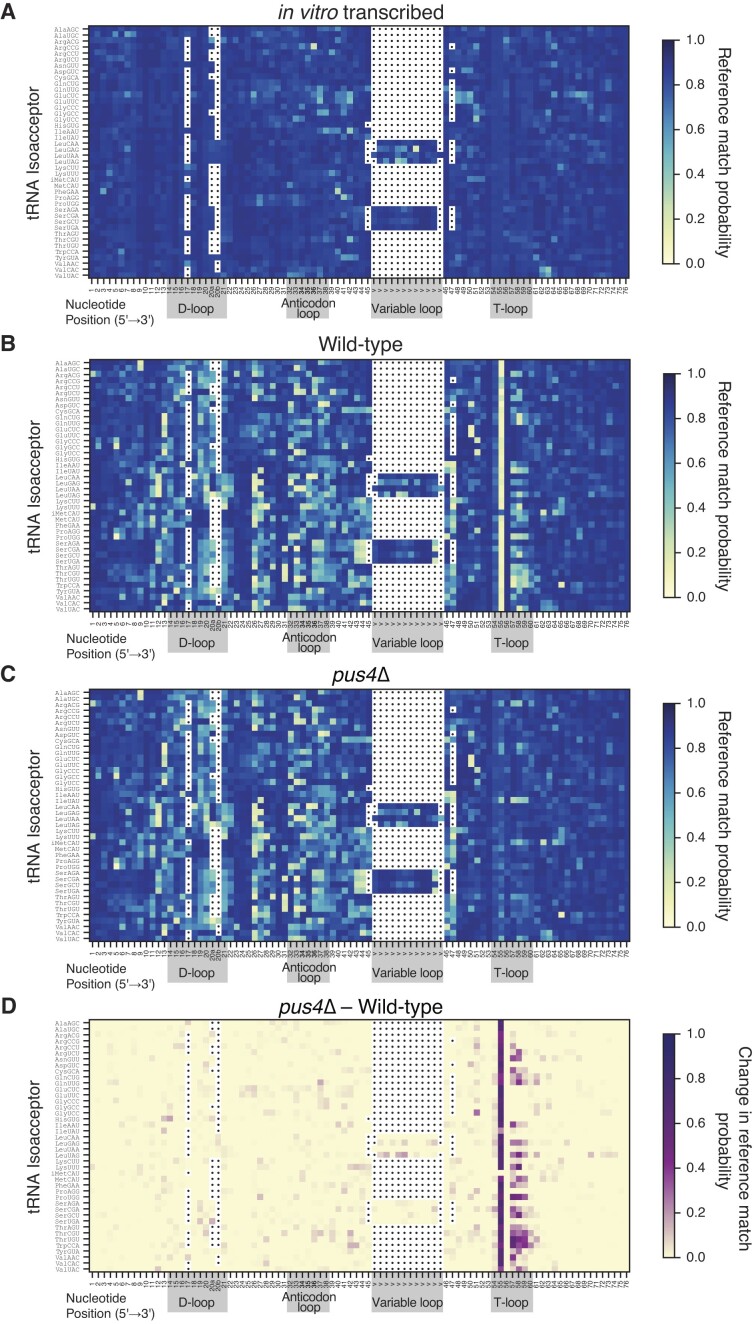
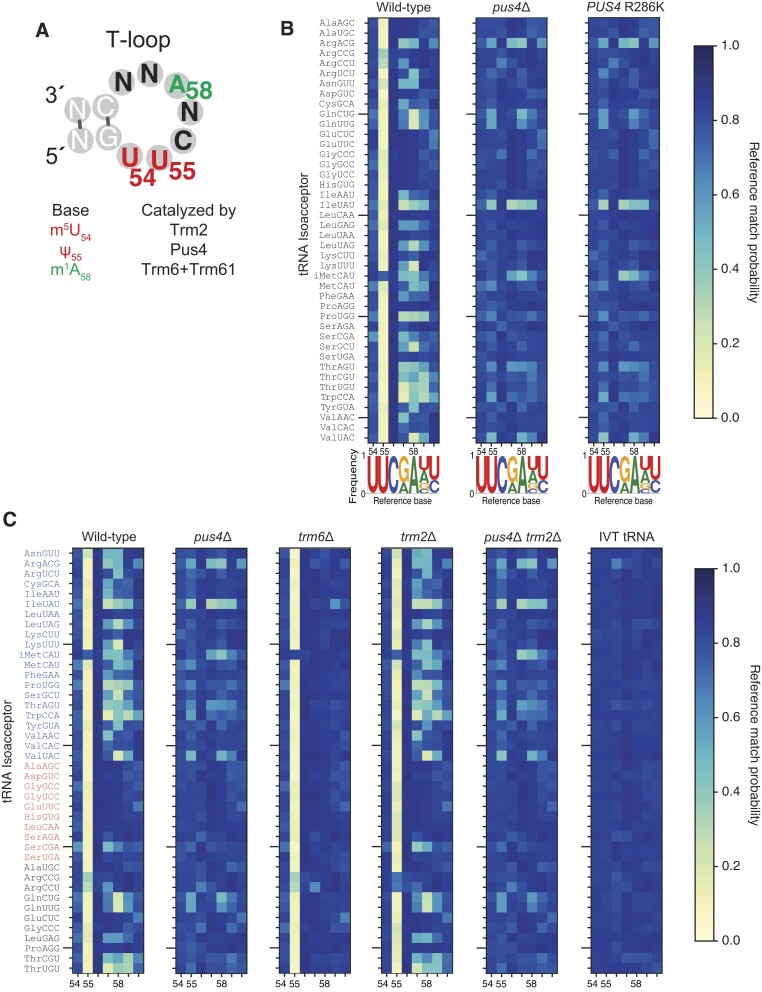
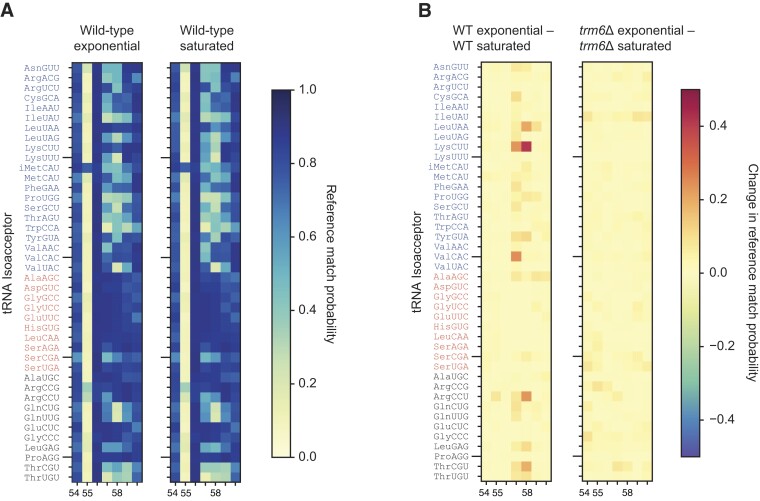
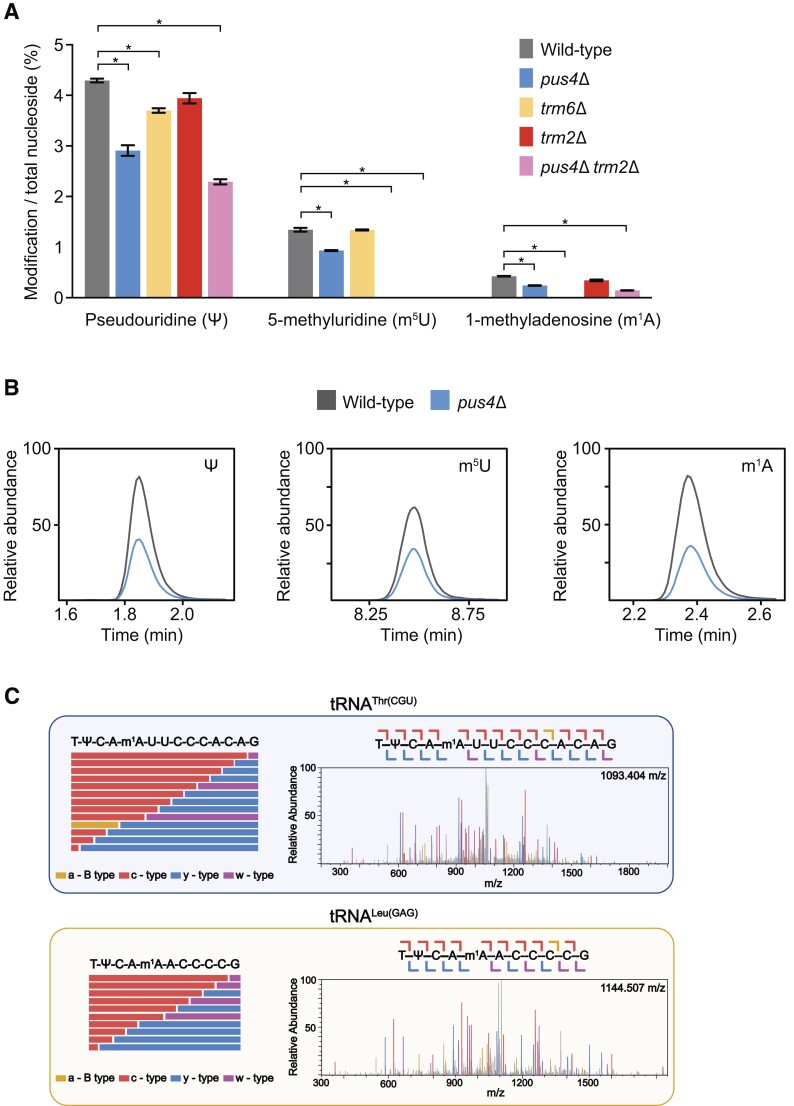
Transfer RNAs (tRNAs) contain dozens of chemical modifications. These modifications are critical for maintaining tRNA tertiary structure and optimizing protein synthesis. Here we advance the use of Nanopore direct RNA-sequencing (DRS) to investigate the synergy between modifications that are known to stabilize tRNA structure. We sequenced the 42 cytosolic tRNA isoacceptors from wild-type yeast and five tRNA-modifying enzyme knockout mutants. These data permitted comprehensive analysis of three neighboring and conserved modifications in T-loops: 5-methyluridine (m5U54), pseudouridine (Ψ55), and 1-methyladenosine (m1A58). Our results were validated using direct measurements of chemical modifications by mass spectrometry. We observed concerted T-loop modification circuits-the potent influence of Ψ55 for subsequent m1A58 modification on more tRNA isoacceptors than previously observed. Growing cells under nutrient depleted conditions also revealed a novel condition-specific increase in m1A58 modification on some tRNAs. A global and isoacceptor-specific classification strategy was developed to predict the status of T-loop modifications from a user-input tRNA DRS dataset, applicable to other conditions and tRNAs in other organisms. These advancements demonstrate how orthogonal technologies combined with genetics enable precise detection of modification landscapes of individual, full-length tRNAs, at transcriptome-scale.
© The Author(s) 2024. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Yoluç Y., Ammann G., Barraud P., Jora M., Limbach P.A., Motorin Y., Marchand V., Tisné C., Borland K., Kellner S.. Instrumental analysis of RNA modifications. Crit. Rev. Biochem. Mol. Biol. 2021; 56:178–204. - PubMed
-
- RajBhandary U.L., Köhrer C.. Early days of tRNA research: discovery, function, purification and sequence analysis. J. Biosci. 2006; 31:439–451. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
