

QSOX2 Deficiency-induced short stature, gastrointestinal dysmotility and immune dysfunction
- PMID: 39341815
- PMCID: PMC11439042
- DOI: 10.1038/s41467-024-52587-w
QSOX2 Deficiency-induced short stature, gastrointestinal dysmotility and immune dysfunction
Abstract
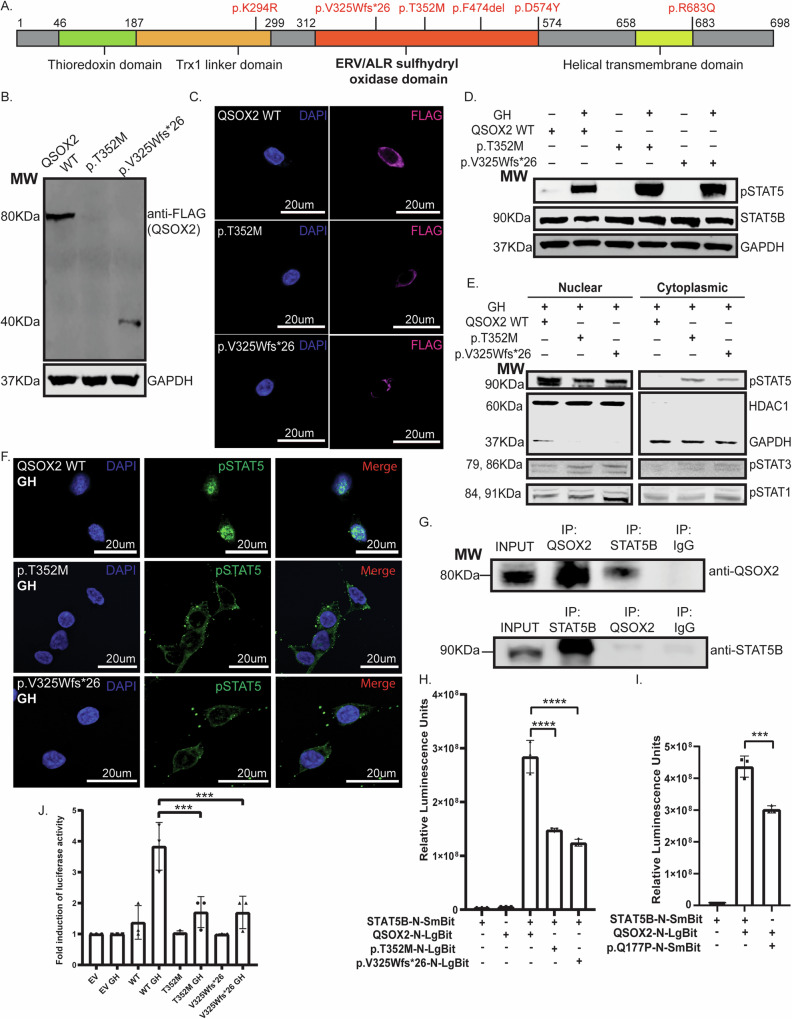
Postnatal growth failure is often attributed to dysregulated somatotropin action, however marked genetic and phenotypic heterogeneity exist. We report five patients from three families who present with short stature, immune dysfunction, atopic eczema and gastrointestinal pathology associated with recessive variants in QSOX2. QSOX2 encodes a nuclear membrane protein linked to disulphide isomerase and oxidoreductase activity. Loss of QSOX2 disrupts Growth hormone-mediated STAT5B nuclear translocation despite enhanced Growth hormone-induced STAT5B phosphorylation. Moreover, patient-derived dermal fibroblasts demonstrate Growth hormone-induced mitochondriopathy and reduced mitochondrial membrane potential. Located at the nuclear membrane, QSOX2 acts as a gatekeeper for regulating stabilisation and import of phosphorylated-STAT5B. Altogether, QSOX2 deficiency modulates human growth by impairing Growth hormone-STAT5B downstream activities and mitochondrial dynamics, which contribute to multi-system dysfunction. Furthermore, our work suggests that therapeutic recombinant insulin-like growth factor-1 may circumvent the Growth hormone-STAT5B dysregulation induced by pathological QSOX2 variants and potentially alleviate organ specific disease.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Murray, P. G., Clayton, P. E. & Chernausek, S. D. A genetic approach to evaluation of short stature of undetermined cause. Lancet Diabetes Endocrinol.6, 564–574 (2018). - PubMed
-
- Vidarsdottir, S. et al. Clinical and biochemical characteristics of a male patient with a novel homozygous STAT5b mutation. J. Clin. Endocrinol. Metab.91, 3482–3485 (2006). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
