

Full-length single-cell BCR sequencing paired with RNA sequencing reveals convergent responses to pneumococcal vaccination
- PMID: 39341987
- PMCID: PMC11438910
- DOI: 10.1038/s42003-024-06823-0
Full-length single-cell BCR sequencing paired with RNA sequencing reveals convergent responses to pneumococcal vaccination
Abstract
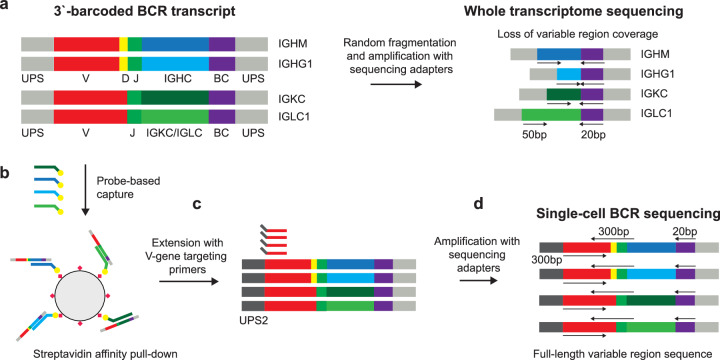
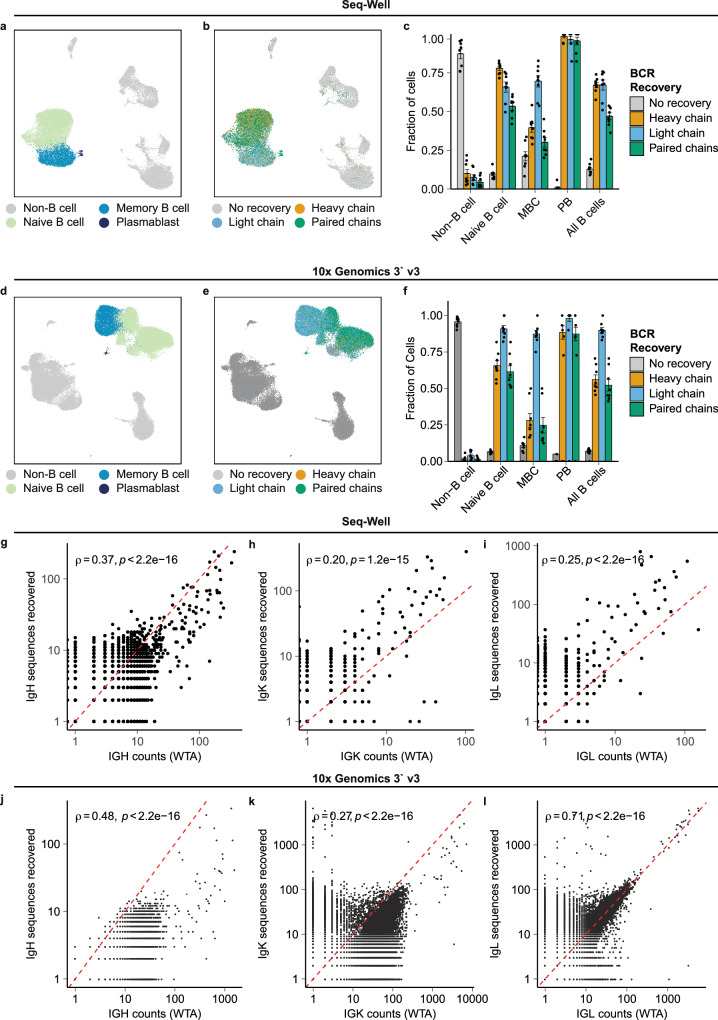
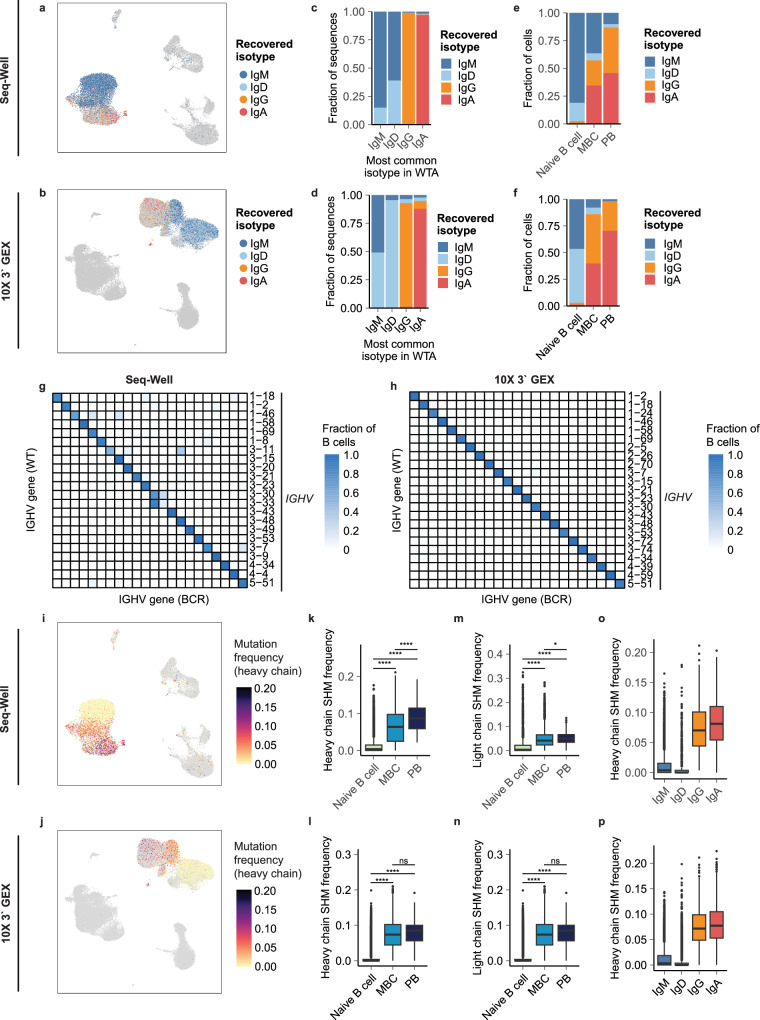
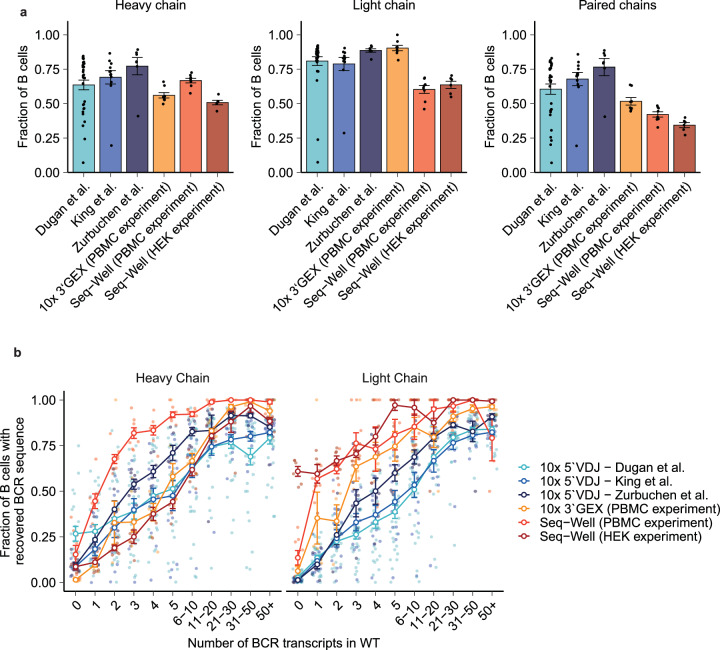
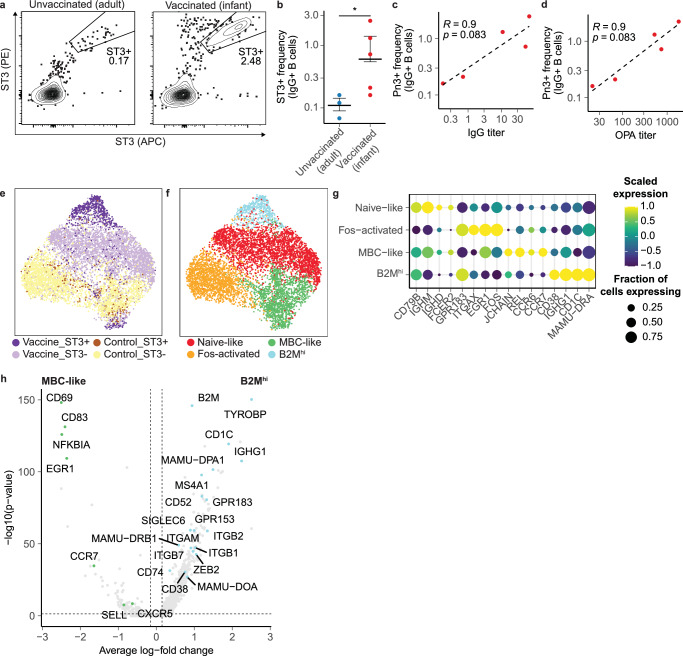
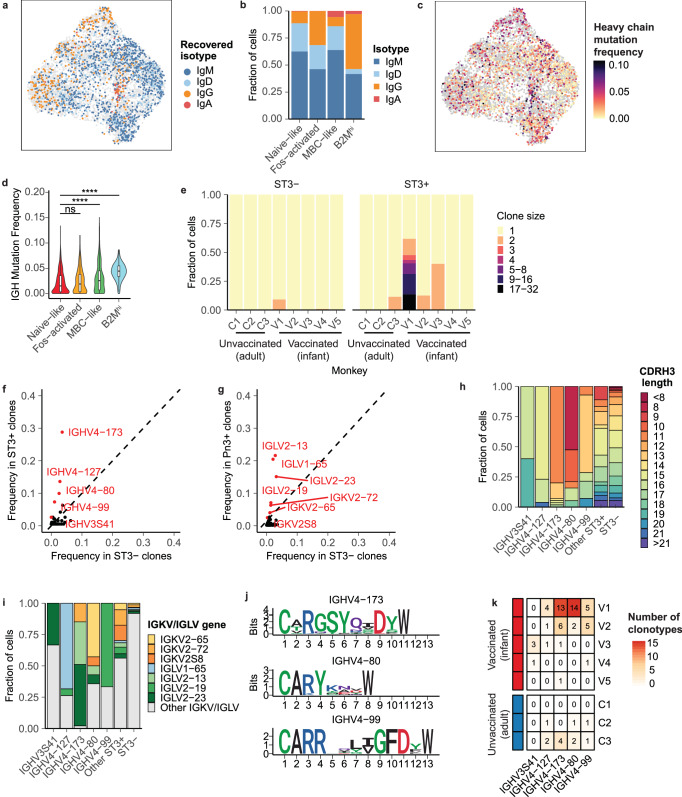
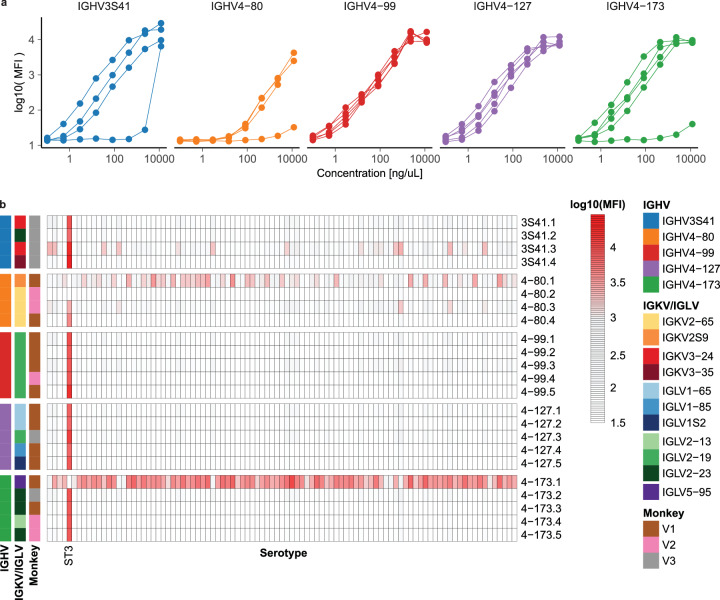
Single-cell RNA sequencing (scRNA-seq) can resolve transcriptional features from individual cells, but scRNA-seq techniques capable of resolving the variable regions of B cell receptors (BCRs) remain limited, especially from widely-used 3'-barcoded libraries. Here, we report a method that can recover paired, full-length variable region sequences of BCRs from 3'-barcoded scRNA-seq libraries. We first verify this method (B3E-seq) can produce accurate, full-length BCR sequences. We then apply this method to profile B cell responses elicited against the capsular polysaccharide of Streptococcus pneumoniae serotype 3 (ST3) by glycoconjugate vaccines in five infant rhesus macaques. We identify BCR features associated with specificity for the ST3 antigen which are present in multiple vaccinated monkeys, indicating a convergent response to vaccination. These results demonstrate the utility of our method to resolve key features of the B cell repertoire and profile antigen-specific responses elicited by vaccination.
© 2024. The Author(s).
Conflict of interest statement
The authors declare the following competing interests: J.C.L. has interests in Sunflower Therapeutics PBC, Honeycomb Biotechnologies, OneCyte Biotechnologies, SQZ Biotech, Alloy Therapeutics, QuantumCyte, Amgen, and Repligen (these interests are reviewed and managed under Massachusetts Institute of Technology’s policies for potential conflicts of interest); in addition, he receives sponsored research support at Massachusetts Institute of Technology from Amgen, the Bill and Melinda Gates Foundation, Biogen, Pfizer, Sartorius, Mott Corp, TurtleTree, Takeda, and Sanofi, and his spouse is an employee of Sunflower Therapeutics PBC. J.K., M.M., S.S., L.J., N.S., I.K., and L.C. are employees of Pfizer and may, as a consequence, be shareholders. Pfizer was involved in the design, analysis, and interpretation of the data in this research study, the writing of this report, and the decision to publish. SUP consults for Mabylon and Buhlmann and conducts a clinical trial for Regeneron. The other authors declare no competing interests.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
