

Complete mitochondrial genome assembly of Juglans regia unveiled its molecular characteristics, genome evolution, and phylogenetic implications
- PMID: 39342114
- PMCID: PMC11439326
- DOI: 10.1186/s12864-024-10818-w
Complete mitochondrial genome assembly of Juglans regia unveiled its molecular characteristics, genome evolution, and phylogenetic implications
Abstract
Background: The Persian walnut (Juglans regia), an economically vital species within the Juglandaceae family, has seen its mitochondrial genome sequenced and assembled in the current study using advanced Illumina and Nanopore sequencing technology.
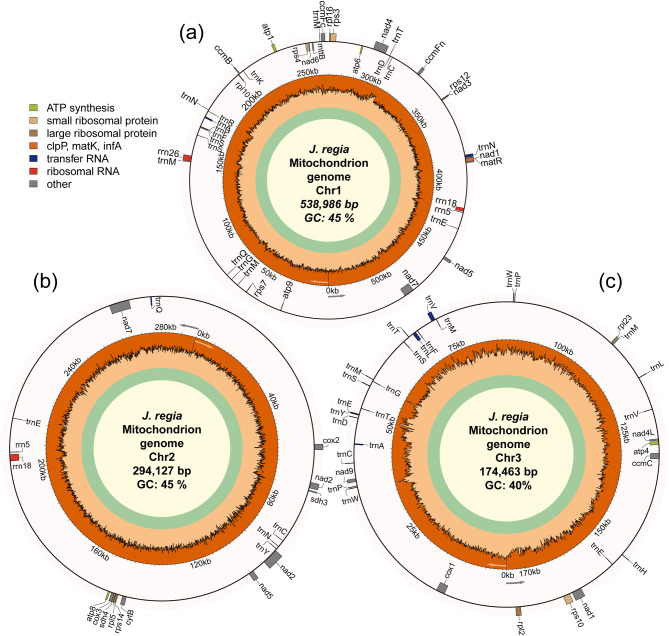
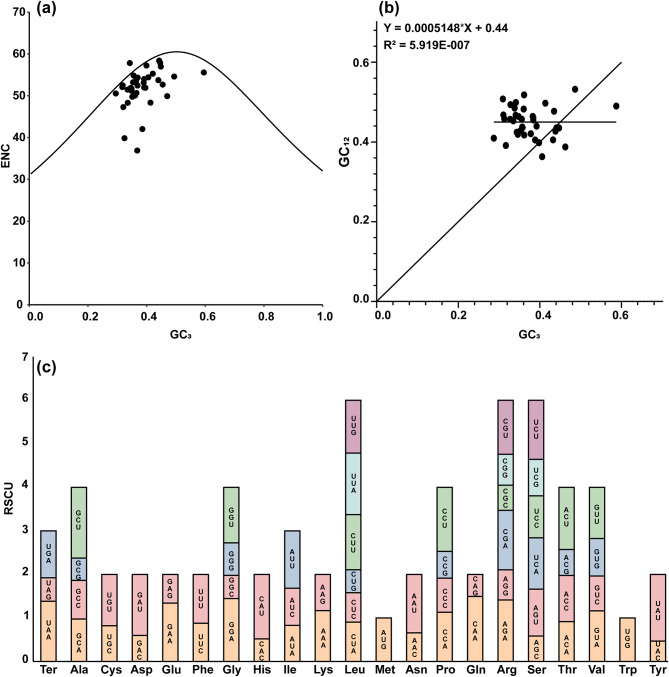
Results: The 1,007,576 bp mitogenome of J. regia consisted of three circular chromosomes with a 44.52% GC content encoding 39 PCGs, 47 tRNA, and five rRNA genes. Extensive repetitive sequences, including 320 SSRs, 512 interspersed, and 83 tandem repeats, were identified, contributing to genomic complexity. The protein-coding sequences (PCGs) favored A/T-ending codons, and the codon usage bias was primarily shaped by selective pressure. Intracellular gene transfer occurred among the mitogenome, chloroplast, and nuclear genomes. Comparative genomic analysis unveiled abundant structure and sequence variation among J. regia and related species. The results of selective pressure analysis indicated that most PCGs underwent purifying selection, whereas the atp4 and ccmB genes had experienced positive selection between many species pairs. In addition, the phylogenetic examination, grounded in mitochondrial genome data, precisely delineated the evolutionary and taxonomic relationships of J. regia and its relatives. We identified a total of 539 RNA editing sites, among which 288 were corroborated by transcriptome sequencing data. Furthermore, expression profiling under temperature stress highlighted the complex regulation pattern of 28 differently expressed PCGs, wherein NADH dehydrogenase and ATP synthase genes might be critical in the mitochondria response to cold stress.
Conclusions: Our results provided valuable molecular resources for understanding the genetic characteristics of J. regia and offered novel perspectives for population genetics and evolutionary studies in Juglans and related woody species.
Keywords: Juglans regia; Comparative analysis; Mitochondrial genome; Temperature stress.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
The authors declare that they have no competing interests.
Figures







References
-
- Song YG, Fragnière Y, Meng HH, Li Y, Bétrisey S, Corrales A, Manchester S, Deng M, Jasińska AK, Văn Sâm H, et al. Global biogeographic synthesis and priority conservation regions of the relict tree family Juglandaceae. J Biogeogr. 2020;47(3):643–57. - DOI
-
- Shiono T, Kusumoto B, Yasuhara M, Kubota Y. Roles of climate niche conservatism and range dynamics in woody plant diversity patterns through the Cenozoic. Glob Ecol Biogeogr. 2018;27(7):865–74. - DOI
MeSH terms
Substances
Grants and funding
- Y23Z619F17/Qinling Hundred Talents Project of Shaanxi Academy of Science
- 32370386/National Natural Science Foundation of China
- 2023-JC-JQ-22/Science Foundation for Distinguished Young Scholars of Shaanxi Province
- 22JHZ005/Basic Research Project of Shaanxi Academy of Fundamental Science
- 2024NC-YBXM-064/Shaanxi Key Research and Development Program
LinkOut - more resources
Full Text Sources
Miscellaneous
