

This is a preprint.
RIPK1 is essential for Herpes Simplex Virus-triggered ZBP1-dependent necroptosis in human cells
- PMID: 39345610
- PMCID: PMC11429907
- DOI: 10.1101/2024.09.17.613393
RIPK1 is essential for Herpes Simplex Virus-triggered ZBP1-dependent necroptosis in human cells
Abstract
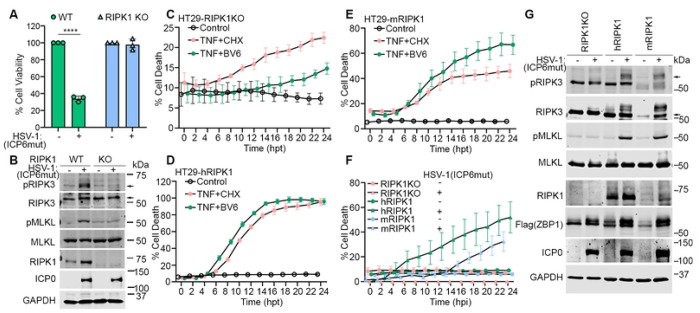
Necroptosis initiated by the host sensor Z-NA Binding Protein-1 (ZBP1) is essential for host defense against a growing number of viruses, including Herpes Simplex Virus-1 (HSV-1). Studies with HSV-1 and other necroptogenic stimuli in murine settings have suggested that ZBP1 triggers necroptosis by directly complexing with the kinase RIPK3. Whether this is also the case in human cells, or whether additional co-factors are needed for ZBP1-mediated necroptosis, is unclear. Here, we show that ZBP1-induced necroptosis in human cells requires RIPK1. We have found that RIPK1 is essential for forming a stable and functional ZBP1-RIPK3 complex in human cells, but is dispensable for the formation of the equivalent murine complex. The RIP Homology Interaction Motif (RHIM) in RIPK3 is responsible for this difference between the two species, because replacing the RHIM in human RIPK3 with the RHIM from murine RIPK3 is sufficient to overcome the requirement for RIPK1 in human cells. These observations describe a critical mechanistic difference between mice and humans in how ZBP1 engages in necroptosis, with important implications for treating human diseases.
Keywords: Cell Death; HSV-1; Necroptosis; RIPK1; RIPK3; ZBP1.
Conflict of interest statement
DECLARATION OF INTERESTS The authors declare no competing interests.
Figures






Similar articles
-
RIPK1 is required for ZBP1-driven necroptosis in human cells.PLoS Biol. 2025 Feb 21;23(2):e3002845. doi: 10.1371/journal.pbio.3002845. eCollection 2025 Feb. PLoS Biol. 2025. PMID: 39982916 Free PMC article.
-
RIPK1 inhibits ZBP1-driven necroptosis during development.Nature. 2016 Dec 1;540(7631):129-133. doi: 10.1038/nature20559. Epub 2016 Nov 7. Nature. 2016. PMID: 27819682
-
Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation.Nature. 2020 Apr;580(7803):391-395. doi: 10.1038/s41586-020-2129-8. Epub 2020 Mar 25. Nature. 2020. PMID: 32296175 Free PMC article.
-
Initiation and execution mechanisms of necroptosis: an overview.Cell Death Differ. 2017 Jul;24(7):1184-1195. doi: 10.1038/cdd.2017.65. Epub 2017 May 12. Cell Death Differ. 2017. PMID: 28498367 Free PMC article. Review.
-
The interplay between human herpes simplex virus infection and the apoptosis and necroptosis cell death pathways.Virol J. 2016 May 6;13:77. doi: 10.1186/s12985-016-0528-0. Virol J. 2016. PMID: 27154074 Free PMC article. Review.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous