

Exploring the potential of structure-based deep learning approaches for T cell receptor design
- PMID: 39348412
- PMCID: PMC11466415
- DOI: 10.1371/journal.pcbi.1012489
Exploring the potential of structure-based deep learning approaches for T cell receptor design
Abstract
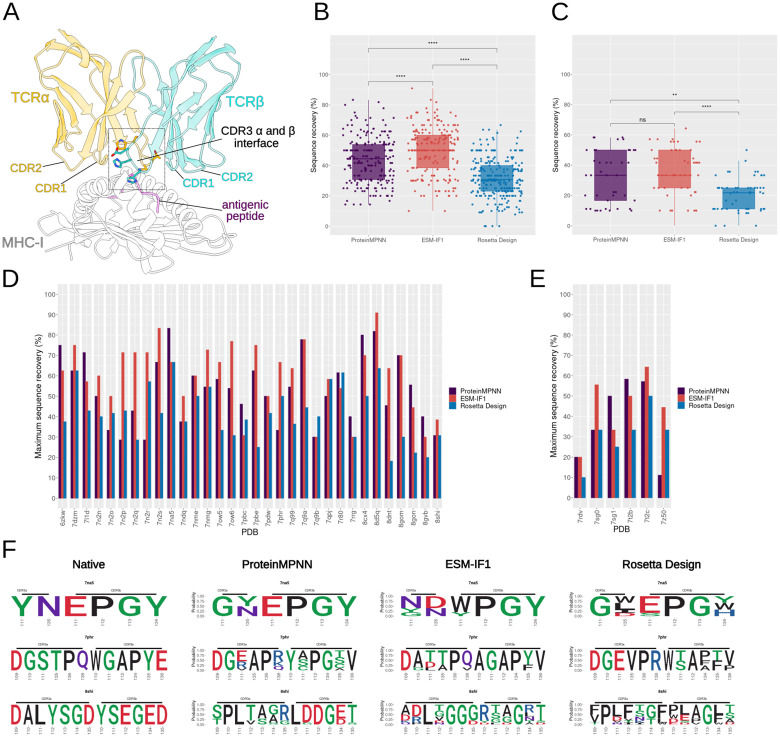
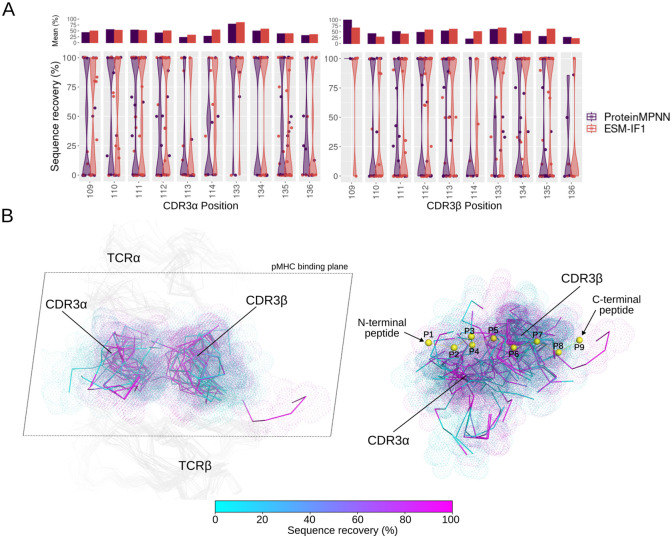
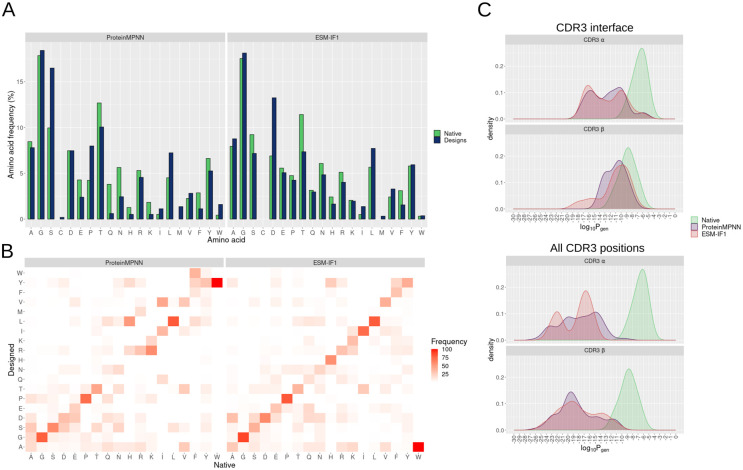
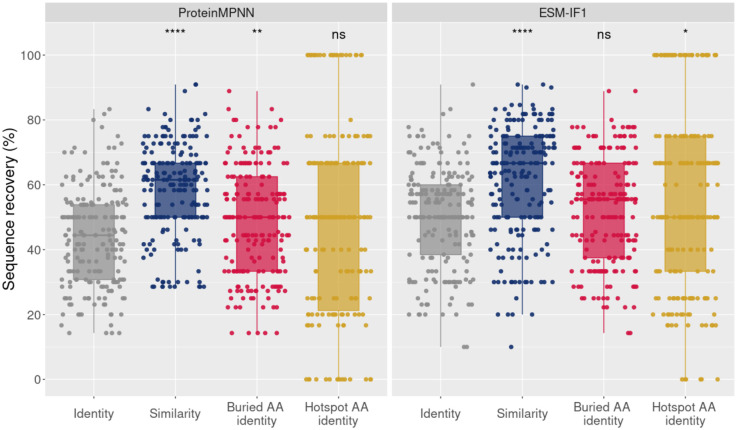
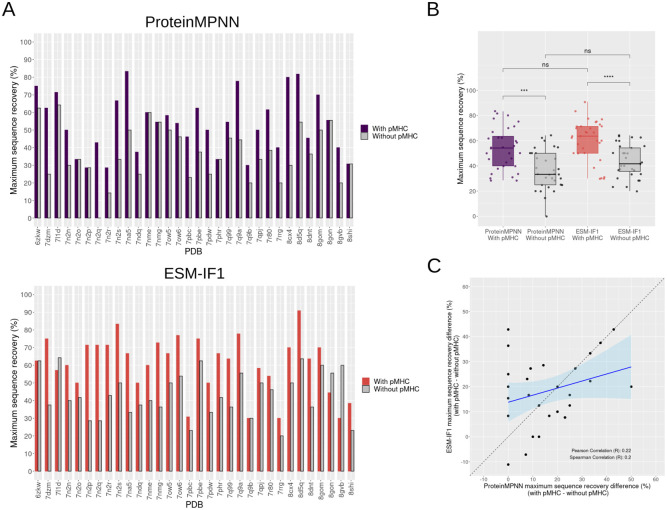
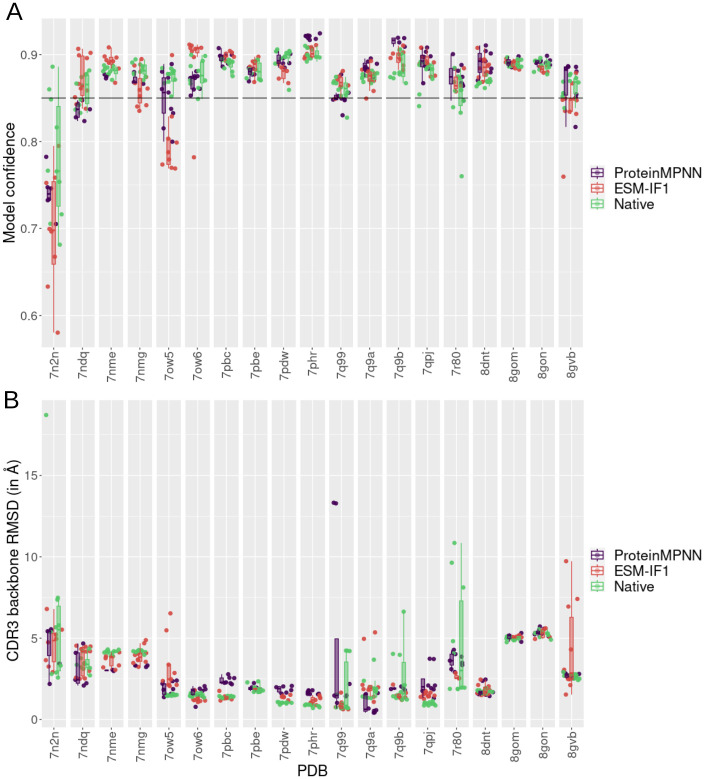
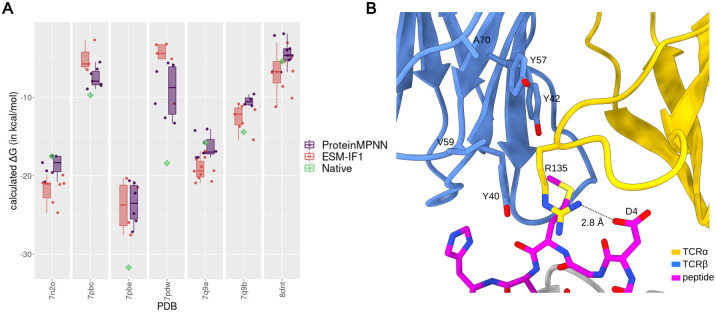
Deep learning methods, trained on the increasing set of available protein 3D structures and sequences, have substantially impacted the protein modeling and design field. These advancements have facilitated the creation of novel proteins, or the optimization of existing ones designed for specific functions, such as binding a target protein. Despite the demonstrated potential of such approaches in designing general protein binders, their application in designing immunotherapeutics remains relatively underexplored. A relevant application is the design of T cell receptors (TCRs). Given the crucial role of T cells in mediating immune responses, redirecting these cells to tumor or infected target cells through the engineering of TCRs has shown promising results in treating diseases, especially cancer. However, the computational design of TCR interactions presents challenges for current physics-based methods, particularly due to the unique natural characteristics of these interfaces, such as low affinity and cross-reactivity. For this reason, in this study, we explored the potential of two structure-based deep learning protein design methods, ProteinMPNN and ESM-IF1, in designing fixed-backbone TCRs for binding target antigenic peptides presented by the MHC through different design scenarios. To evaluate TCR designs, we employed a comprehensive set of sequence- and structure-based metrics, highlighting the benefits of these methods in comparison to classical physics-based design methods and identifying deficiencies for improvement.
Copyright: © 2024 Ribeiro-Filho et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Update of
-
Exploring the Potential of Structure-Based Deep Learning Approaches for T cell Receptor Design.bioRxiv [Preprint]. 2024 Apr 24:2024.04.19.590222. doi: 10.1101/2024.04.19.590222. bioRxiv. 2024. Update in: PLoS Comput Biol. 2024 Sep 30;20(9):e1012489. doi: 10.1371/journal.pcbi.1012489. PMID: 38712216 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
