

Valid inference for machine learning-assisted genome-wide association studies
- PMID: 39349818
- PMCID: PMC11972620
- DOI: 10.1038/s41588-024-01934-0
Valid inference for machine learning-assisted genome-wide association studies
Abstract
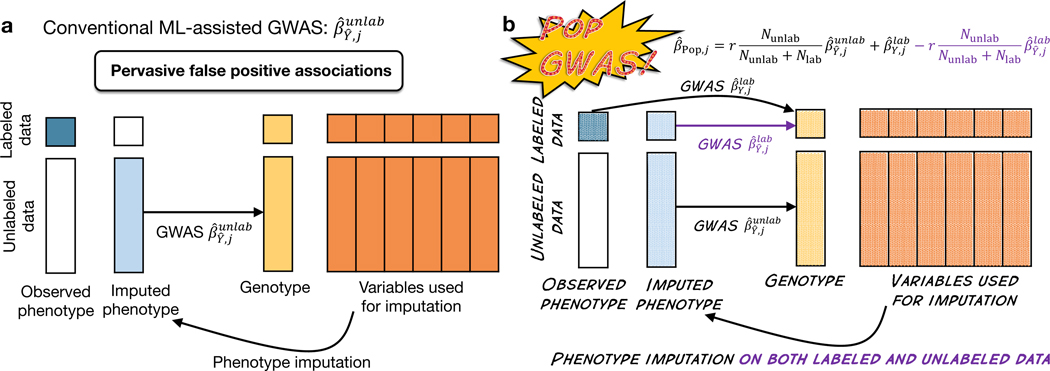
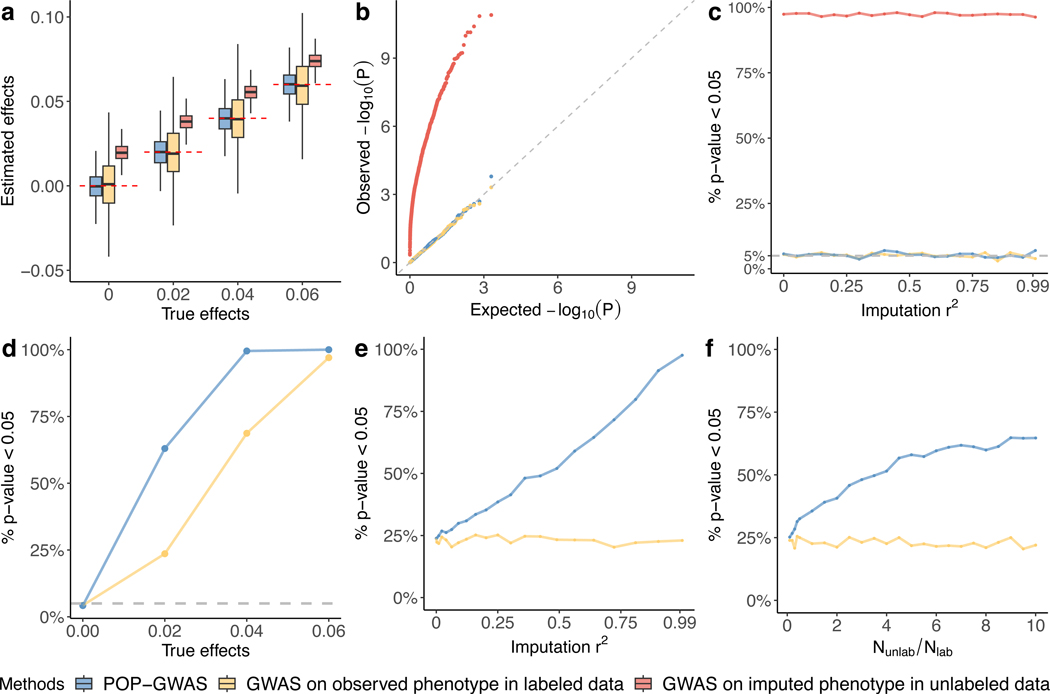
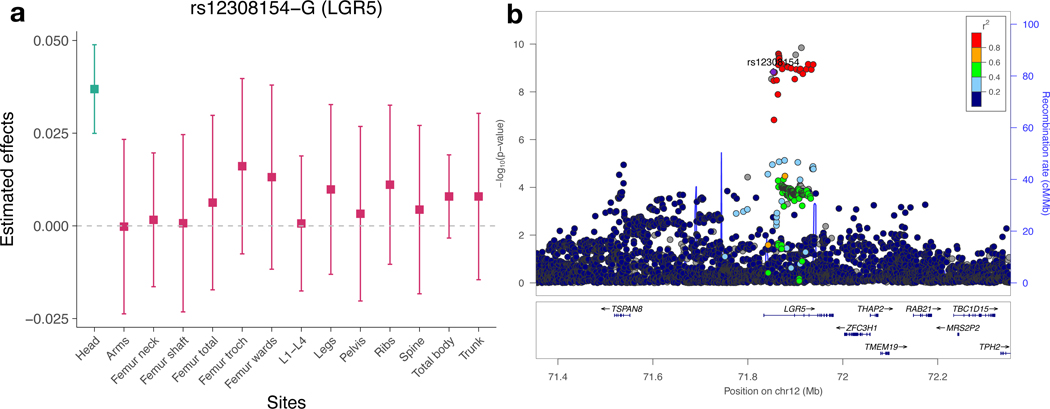
Machine learning (ML) has become increasingly popular in almost all scientific disciplines, including human genetics. Owing to challenges related to sample collection and precise phenotyping, ML-assisted genome-wide association study (GWAS), which uses sophisticated ML techniques to impute phenotypes and then performs GWAS on the imputed outcomes, have become increasingly common in complex trait genetics research. However, the validity of ML-assisted GWAS associations has not been carefully evaluated. Here, we report pervasive risks for false-positive associations in ML-assisted GWAS and introduce Post-Prediction GWAS (POP-GWAS), a statistical framework that redesigns GWAS on ML-imputed outcomes. POP-GWAS ensures valid and powerful statistical inference irrespective of imputation quality and choice of algorithm, requiring only GWAS summary statistics as input. We employed POP-GWAS to perform a GWAS of bone mineral density derived from dual-energy X-ray absorptiometry imaging at 14 skeletal sites, identifying 89 new loci and revealing skeletal site-specific genetic architecture. Our framework offers a robust analytic solution for future ML-assisted GWAS.
© 2024. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures



References
-
- Uffelmann E et al. Genome-wide association studies. Nature Reviews Methods Primers 1, 59 (2021).
-
- Cosentino J et al. Inference of chronic obstructive pulmonary disease with deep learning on raw spirograms identifies new genetic loci and improves risk models. Nature Genetics, 1–9 (2023). - PubMed
Methods-only references
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
