

Genomic and transcriptomic features of androgen receptor signaling inhibitor resistance in metastatic castration-resistant prostate cancer
- PMID: 39352383
- PMCID: PMC11444163
- DOI: 10.1172/JCI178604
Genomic and transcriptomic features of androgen receptor signaling inhibitor resistance in metastatic castration-resistant prostate cancer
Abstract
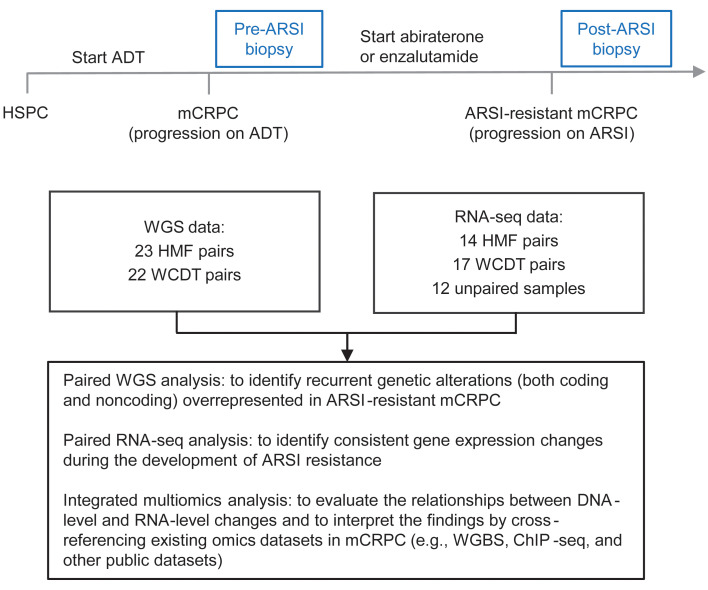
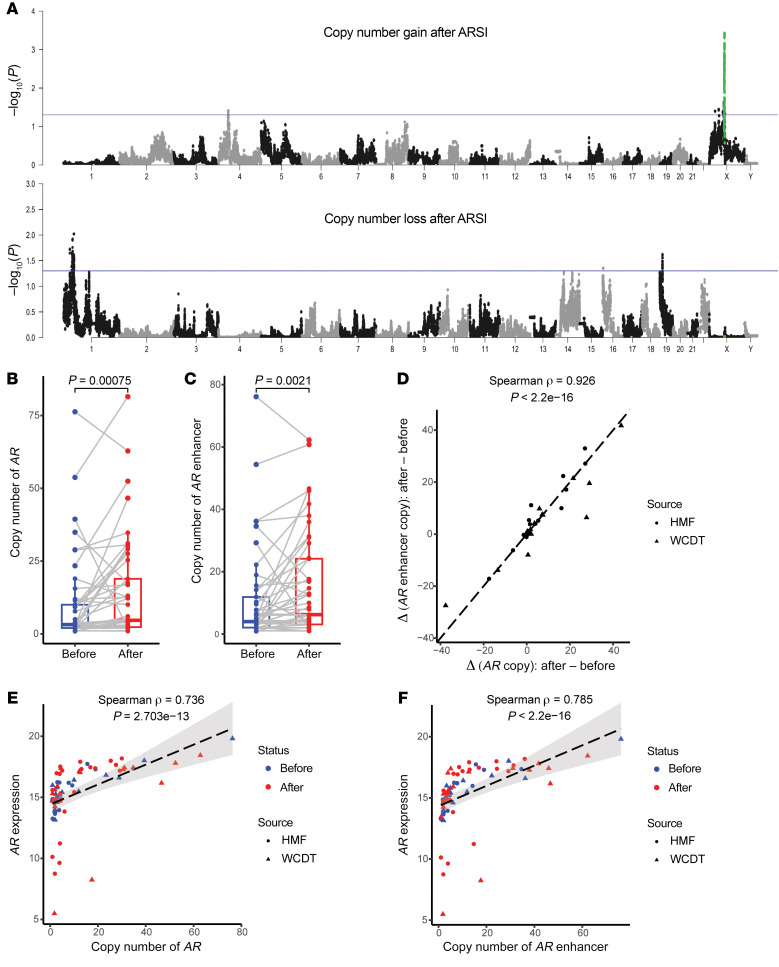
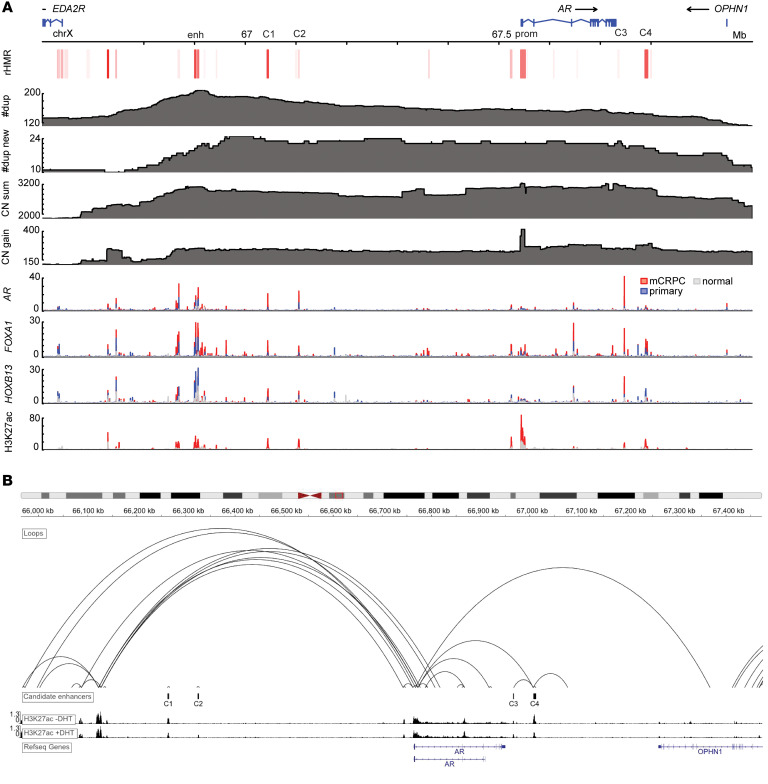
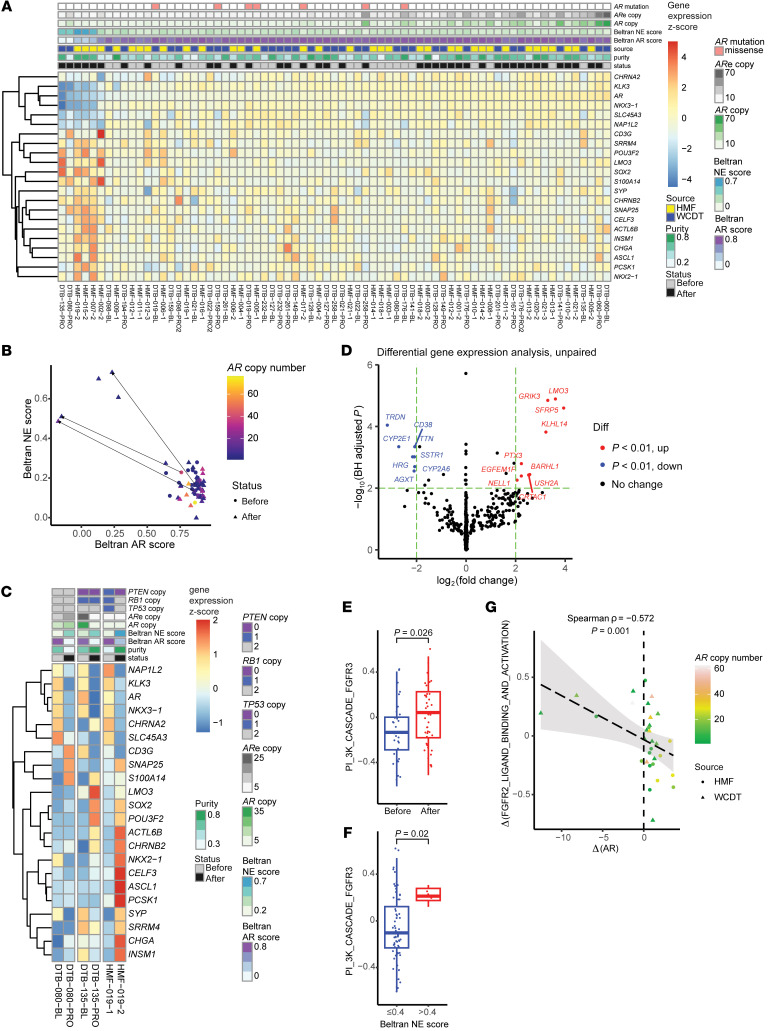
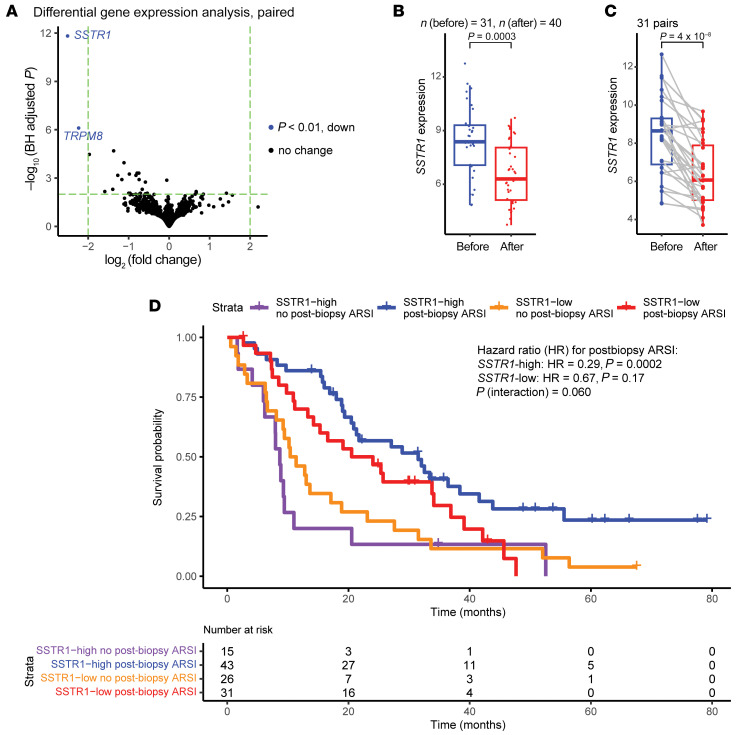
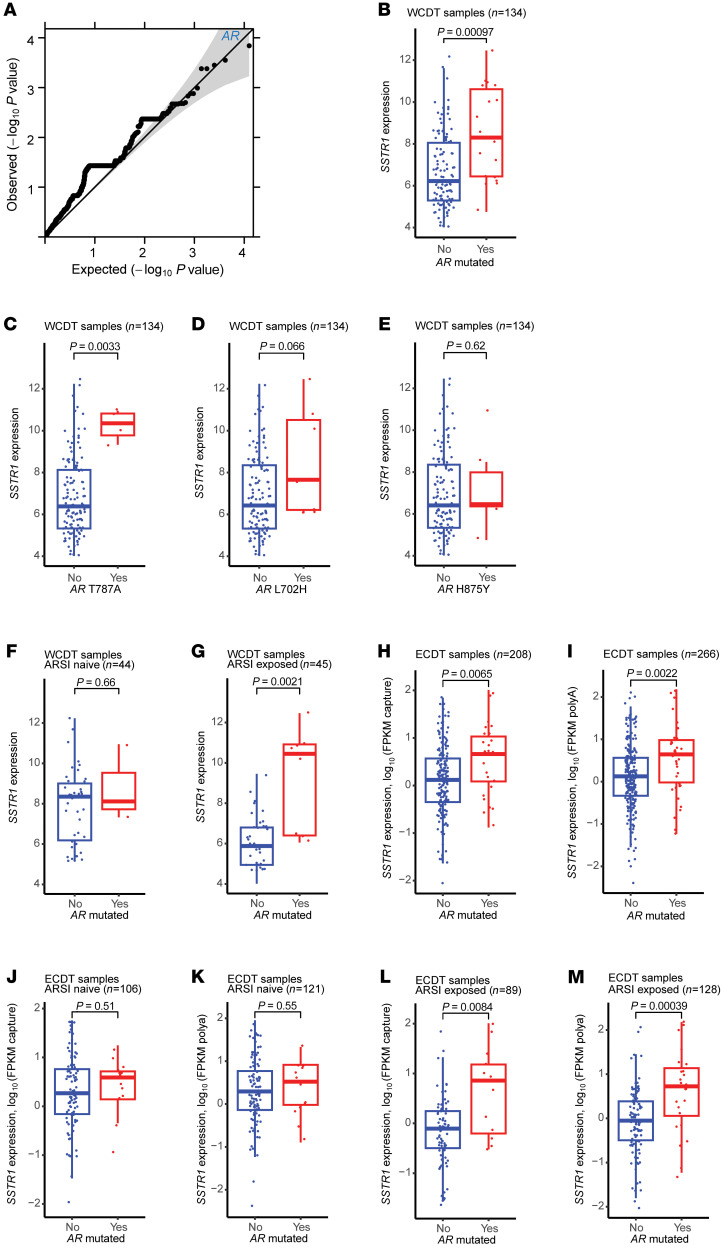
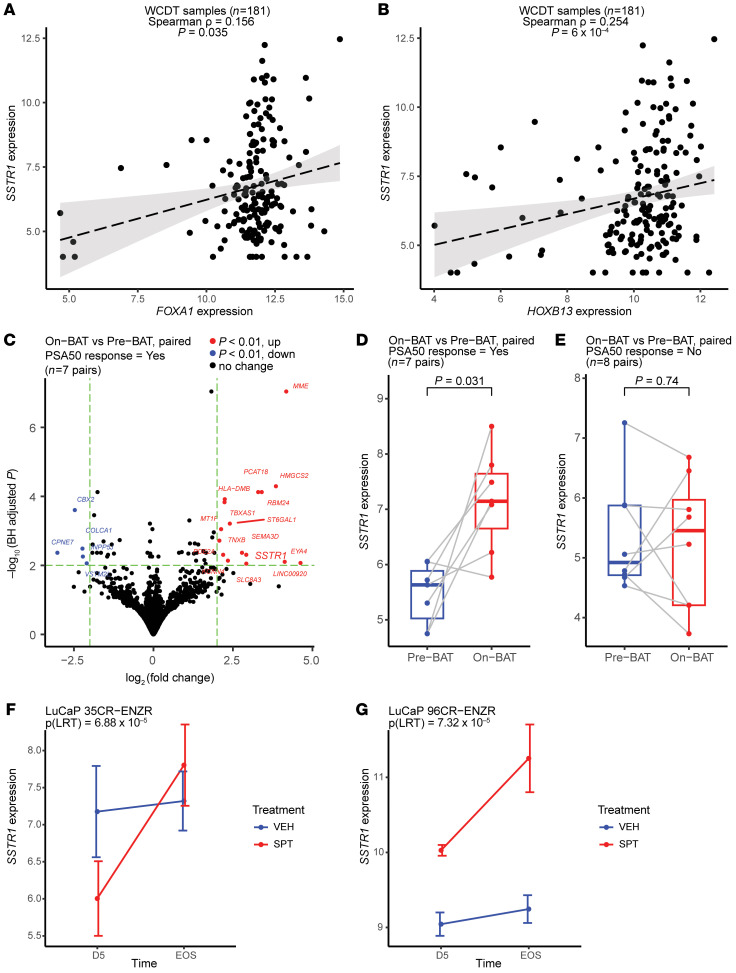
BACKGROUNDAndrogen receptor signaling inhibitors (ARSIs) have improved outcomes for patients with metastatic castration-resistant prostate cancer (mCRPC), but their clinical benefit is limited by treatment resistance.METHODSTo investigate the mechanisms of ARSI resistance, we analyzed the whole-genome (n = 45) and transcriptome (n = 31) sequencing data generated from paired metastatic biopsies obtained before initiation of first-line ARSI therapy for mCRPC and after radiographic disease progression. We investigated the effects of genetic and pharmacologic modulation of SSTR1 in 22Rv1 cells, a representative mCRPC cell line.RESULTSWe confirmed the predominant role of tumor genetic alterations converging on augmenting androgen receptor (AR) signaling and the increased transcriptional heterogeneity and lineage plasticity during the emergence of ARSI resistance. We further identified amplifications involving a putative enhancer downstream of the AR and transcriptional downregulation of SSTR1, encoding somatostatin receptor 1, in ARSI-resistant tumors. We found that patients with SSTR1-low mCRPC tumors derived less benefit from subsequent ARSI therapy in a retrospective cohort. We showed that SSTR1 was antiproliferative in 22Rv1 cells and that the FDA-approved drug pasireotide suppressed 22Rv1 cell proliferation.CONCLUSIONOur findings expand the knowledge of ARSI resistance and point out actionable next steps, exemplified by potentially targeting SSTR1, to improve patient outcomes.FUNDINGNational Cancer Institute (NCI), NIH; Prostate Cancer Foundation; Conquer Cancer, American Society of Clinical Oncology Foundation; UCSF Benioff Initiative for Prostate Cancer Research; Netherlands Cancer Institute.
Keywords: Oncology; Prostate cancer.
Conflict of interest statement
Figures
References
-
- Gillessen S, et al. Management of patients with advanced prostate cancer. Part I: Intermediate-/high-risk and locally advanced disease, biochemical relapse, and side effects of hormonal treatment: Report of the Advanced Prostate Cancer Consensus Conference 2022. Eur Urol. 2023;83(3):267–293. - PMC - PubMed
