

Endothelial YAP/TAZ activation promotes atherosclerosis in a mouse model of Hutchinson-Gilford progeria syndrome
- PMID: 39352768
- PMCID: PMC11563688
- DOI: 10.1172/JCI173448
Endothelial YAP/TAZ activation promotes atherosclerosis in a mouse model of Hutchinson-Gilford progeria syndrome
Abstract
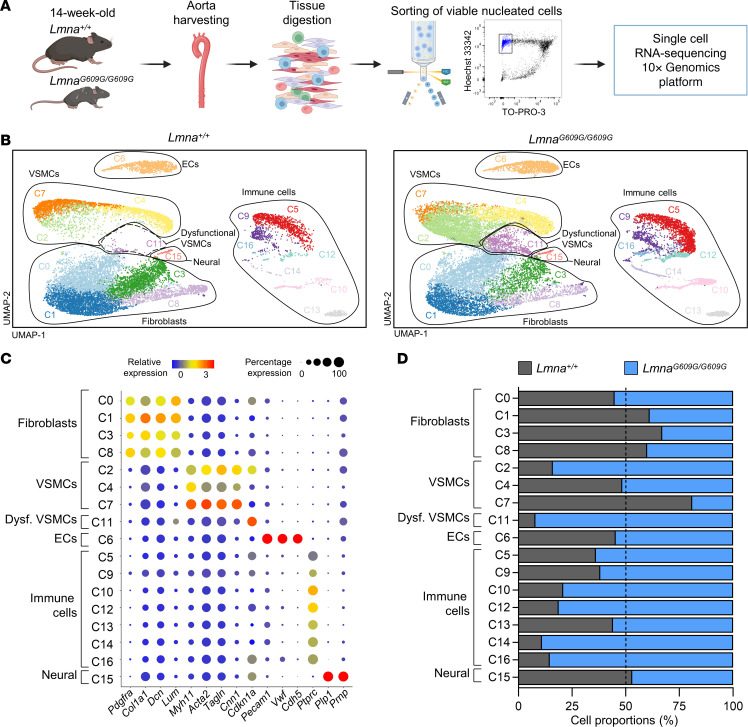
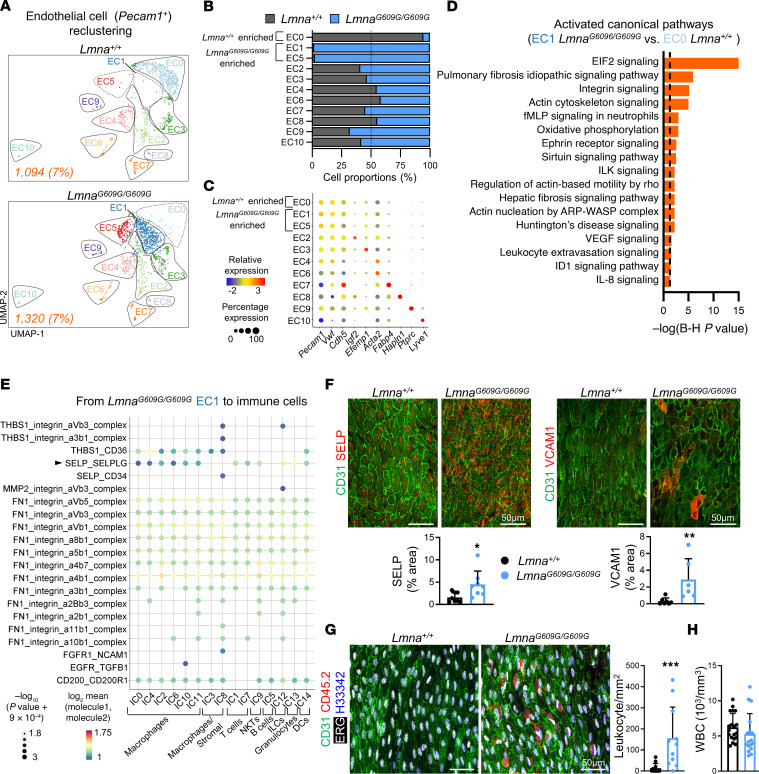
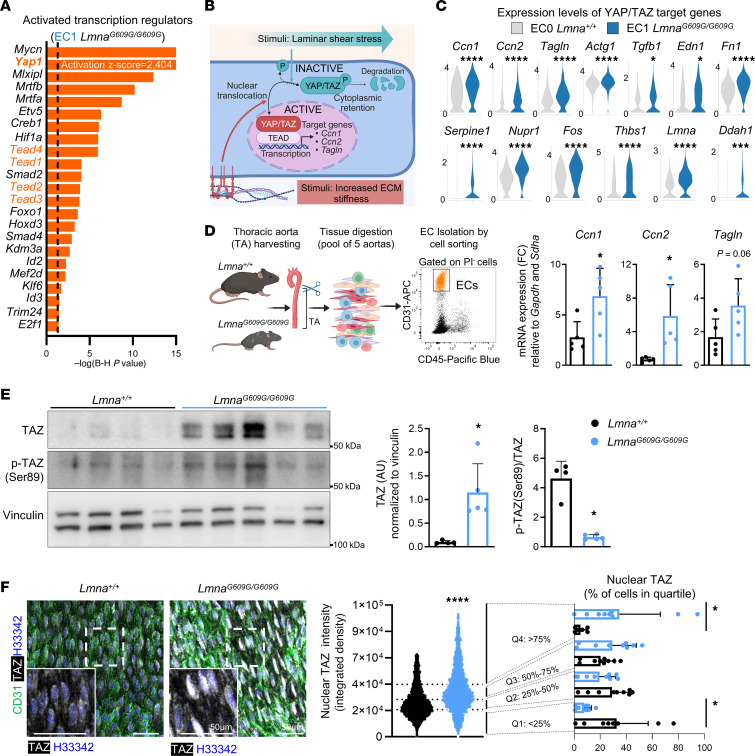
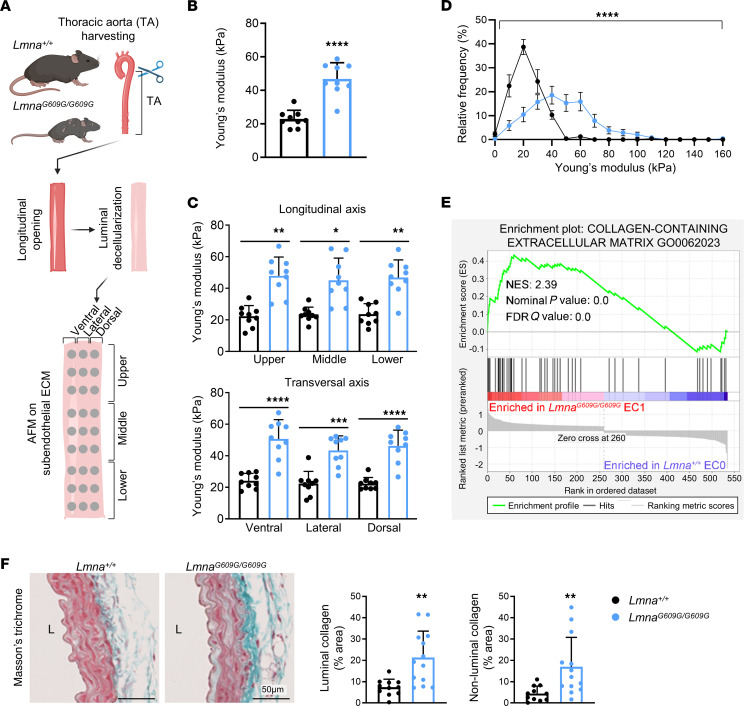
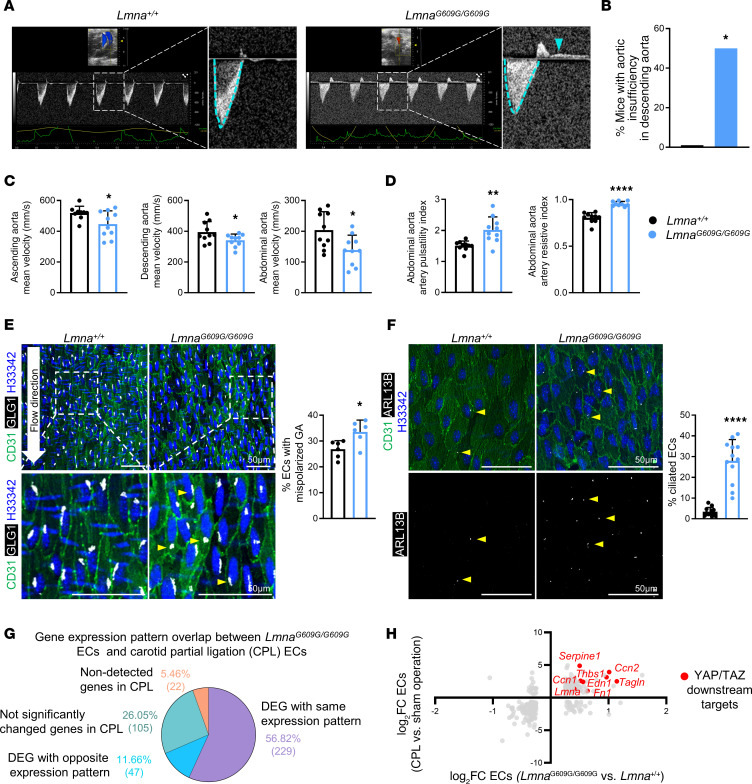
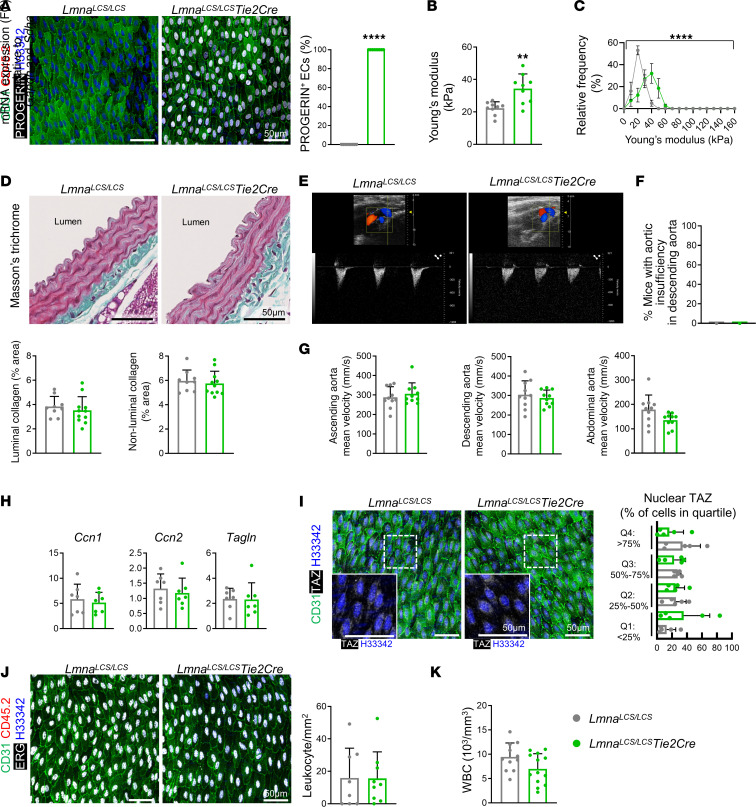
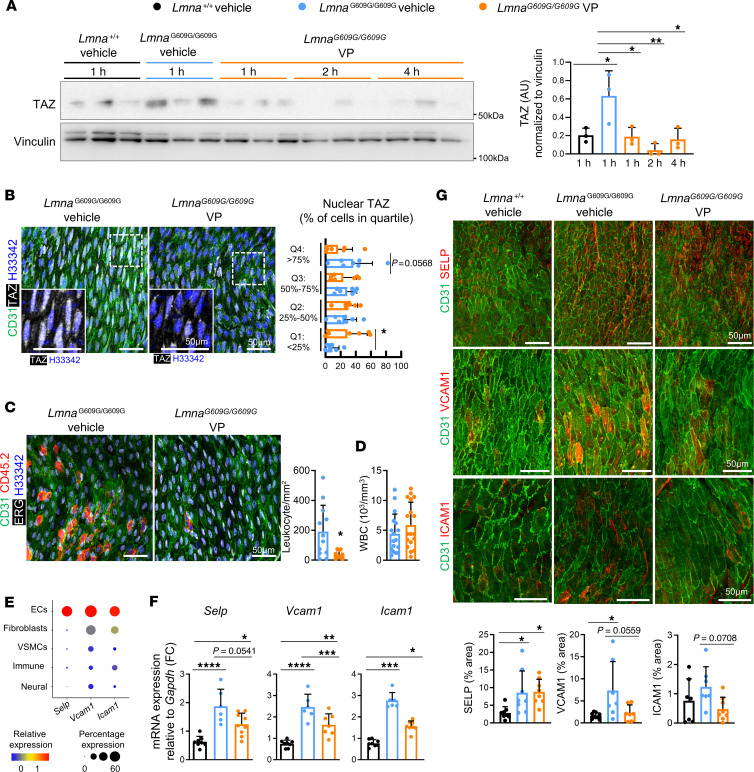
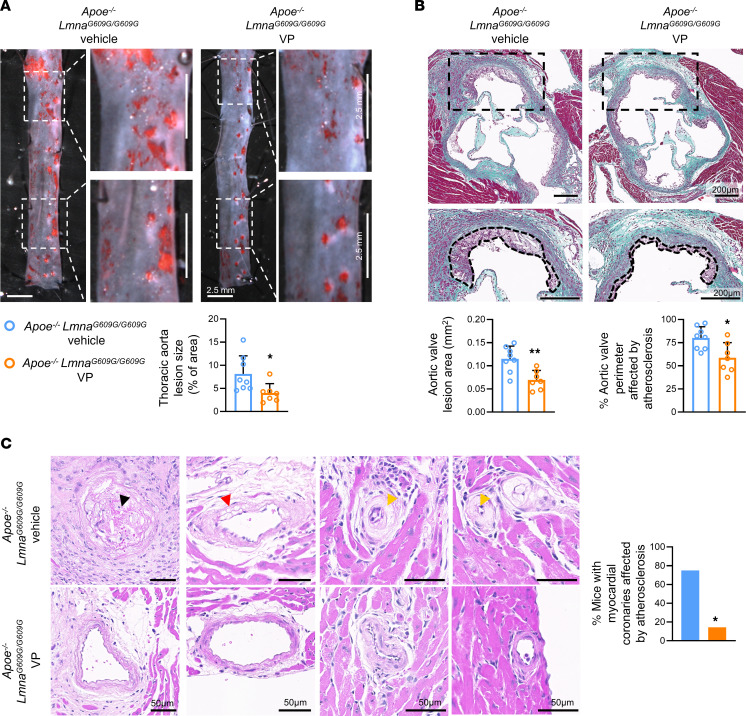
Hutchinson-Gilford progeria syndrome (HGPS) is an extremely rare disease caused by the expression of progerin, an aberrant protein produced by a point mutation in the LMNA gene. HGPS patients show accelerated aging and die prematurely mainly from complications of atherosclerosis such as myocardial infarction, heart failure, or stroke. However, the mechanisms underlying HGPS vascular pathology remain ill-defined. We used single-cell RNA sequencing to characterize the aorta in progerin-expressing LmnaG609G/G609G mice and wild-type controls, with a special focus on endothelial cells (ECs). HGPS ECs showed gene expression changes associated with extracellular matrix alterations, increased leukocyte extravasation, and activation of the yes-associated protein 1/transcriptional activator with PDZ-binding domain (YAP/TAZ) mechanosensing pathway, all validated by different techniques. Atomic force microscopy experiments demonstrated stiffer subendothelial extracellular matrix in progeroid aortae, and ultrasound assessment of live HGPS mice revealed disturbed aortic blood flow, both key inducers of the YAP/TAZ pathway in ECs. YAP/TAZ inhibition with verteporfin reduced leukocyte accumulation in the aortic intimal layer and decreased atherosclerosis burden in progeroid mice. Our findings identify endothelial YAP/TAZ signaling as a key mechanism of HGPS vascular disease and open a new avenue for the development of YAP/TAZ-targeting drugs to ameliorate progerin-induced atherosclerosis.
Keywords: Aging; Atherosclerosis; Cardiovascular disease; Endothelial cells; Vascular biology.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
