

Microbiome-metabolite linkages drive greenhouse gas dynamics over a permafrost thaw gradient
- PMID: 39354152
- PMCID: PMC11522005
- DOI: 10.1038/s41564-024-01800-z
Microbiome-metabolite linkages drive greenhouse gas dynamics over a permafrost thaw gradient
Abstract
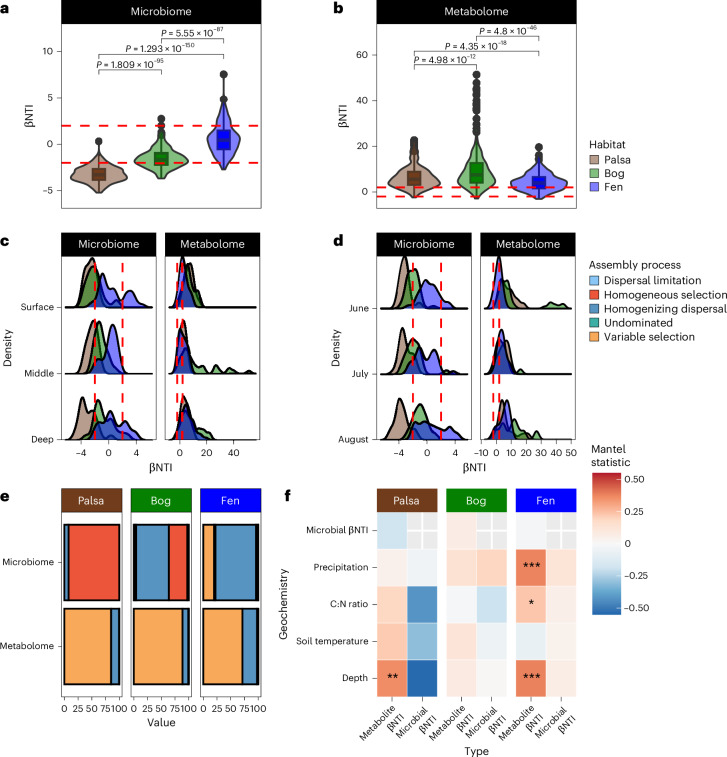
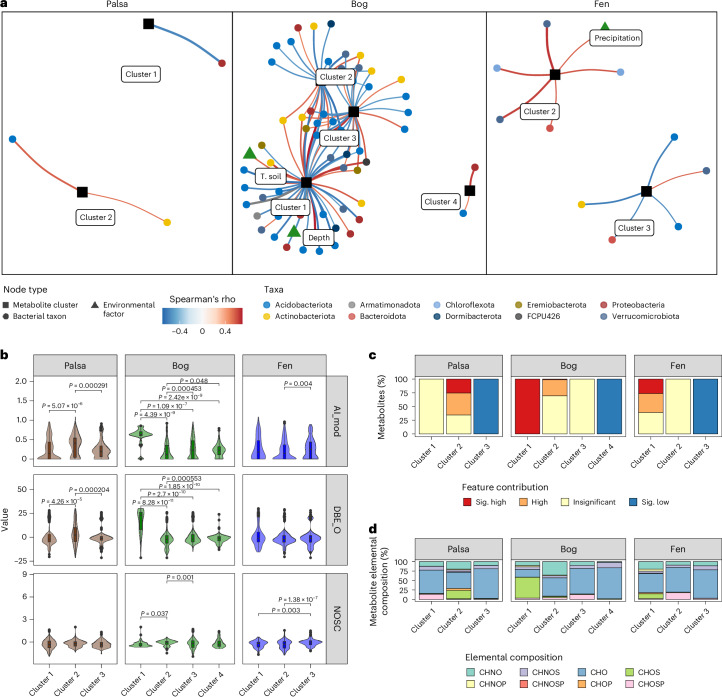
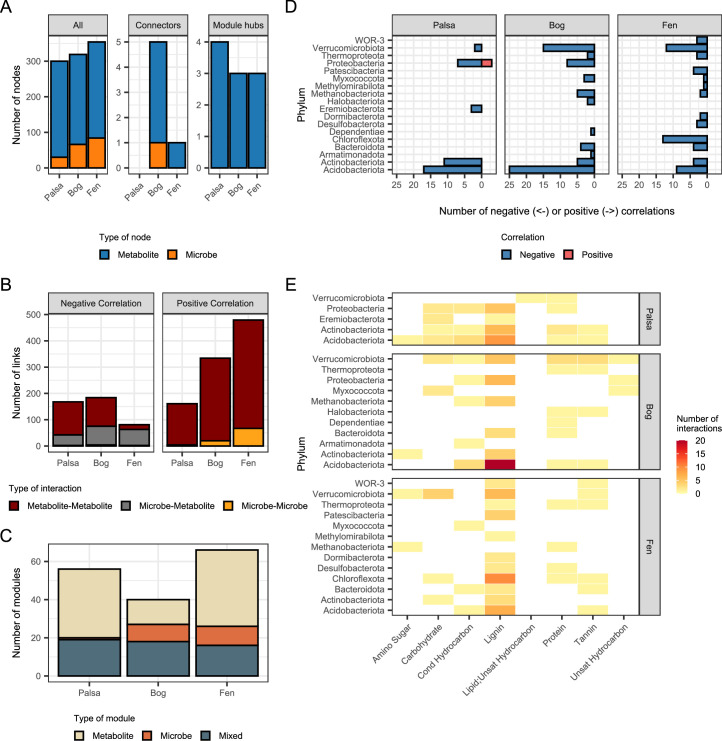
Interactions between microbiomes and metabolites play crucial roles in the environment, yet how these interactions drive greenhouse gas emissions during ecosystem changes remains unclear. Here we analysed microbial and metabolite composition across a permafrost thaw gradient in Stordalen Mire, Sweden, using paired genome-resolved metagenomics and high-resolution Fourier transform ion cyclotron resonance mass spectrometry guided by principles from community assembly theory to test whether microorganisms and metabolites show concordant responses to changing drivers. Our analysis revealed divergence between the inferred microbial versus metabolite assembly processes, suggesting distinct responses to the same selective pressures. This contradicts common assumptions in trait-based microbial models and highlights the limitations of measuring microbial community-level data alone. Furthermore, feature-scale analysis revealed connections between microbial taxa, metabolites and observed CO2 and CH4 porewater variations. Our study showcases insights gained by using feature-level data and microorganism-metabolite interactions to better understand metabolic processes that drive greenhouse gas emissions during ecosystem changes.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures







References
-
- Ma, L. et al. A globally robust relationship between water table decline, subsidence rate, and carbon release from peatlands. Commun. Earth Environ.3, 254 (2022).
-
- Woodcroft, B. J. et al. Genome-centric view of carbon processing in thawing permafrost. Nature560, 49–54 (2018). - PubMed
-
- Shah, R. M. et al. Omics-based ecosurveillance uncovers the influence of estuarine macrophytes on sediment microbial function and metabolic redundancy in a tropical ecosystem. Sci. Total Environ.809, 151175 (2022). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
