

Increased frequency of repeat expansion mutations across different populations
- PMID: 39354197
- PMCID: PMC11564083
- DOI: 10.1038/s41591-024-03190-5
Increased frequency of repeat expansion mutations across different populations
Abstract
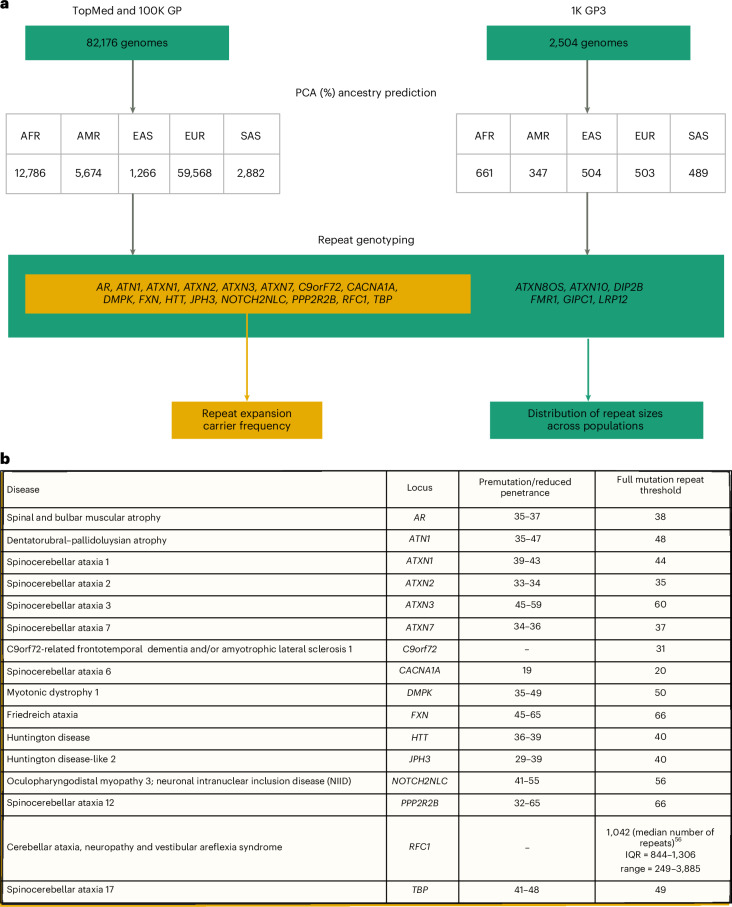
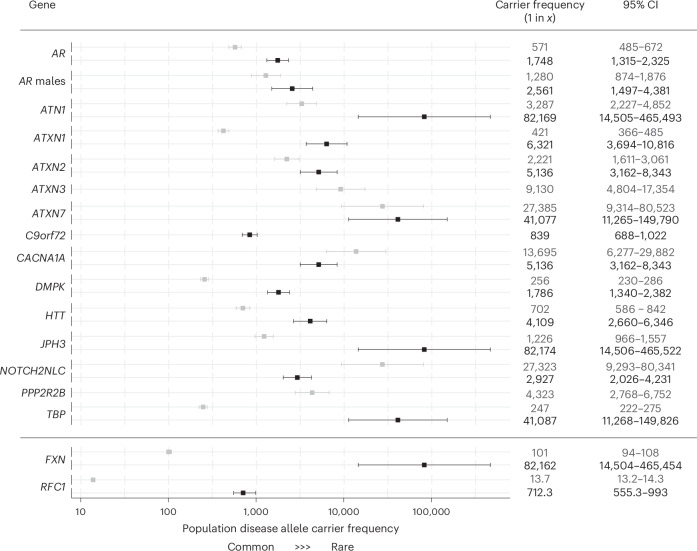
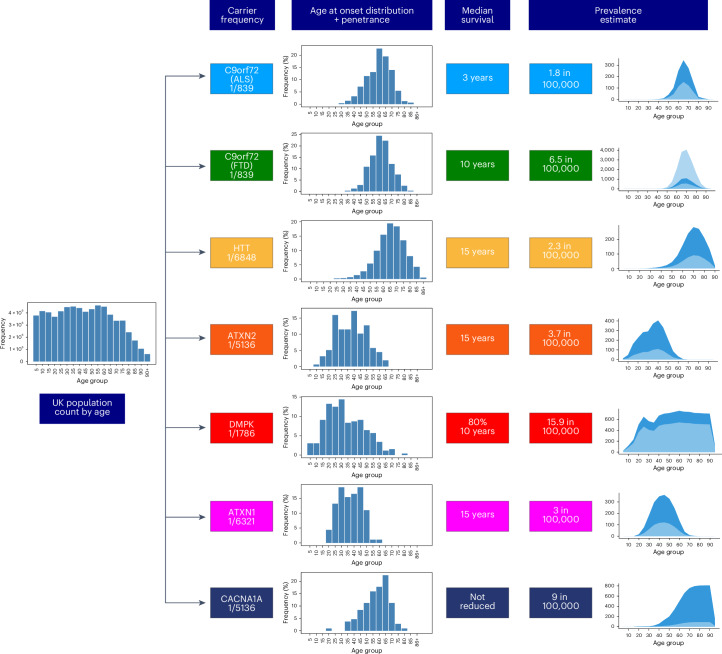
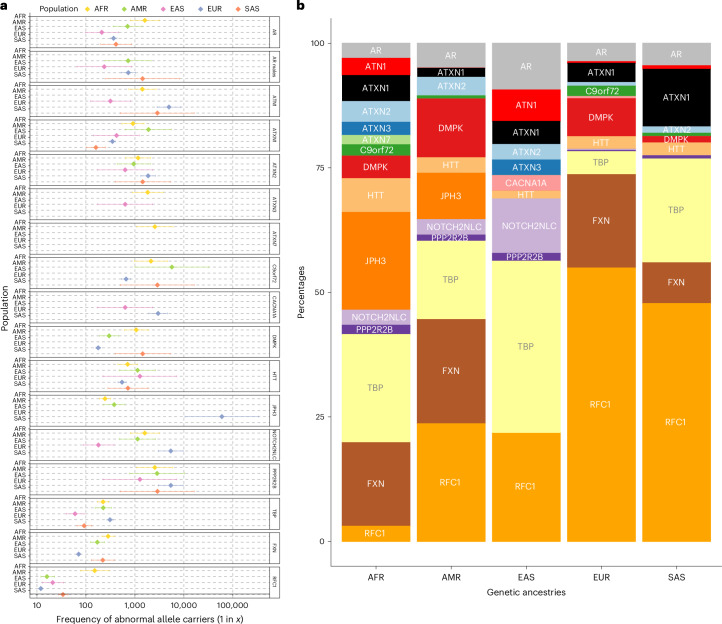
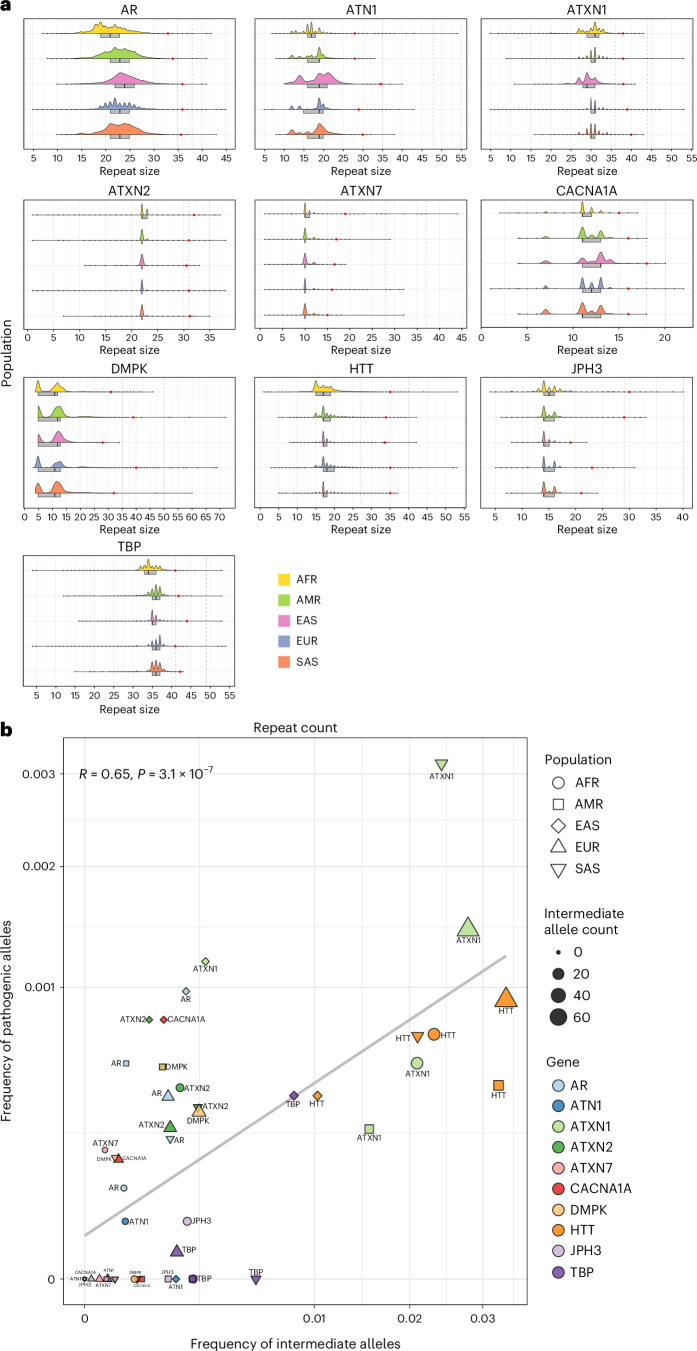
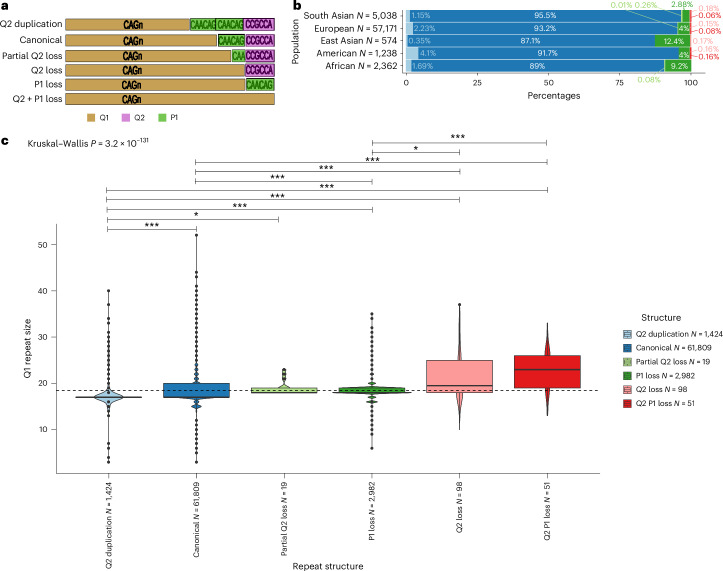
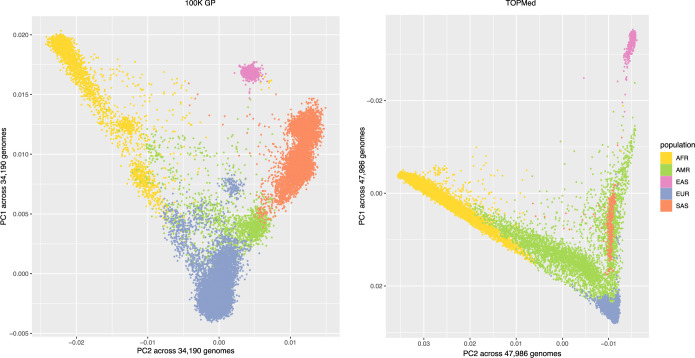
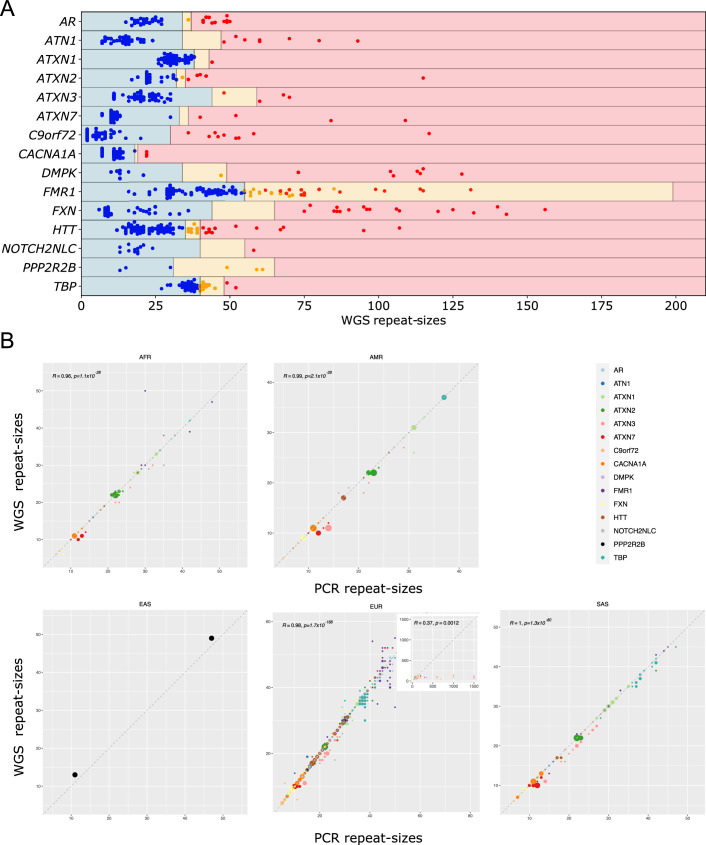
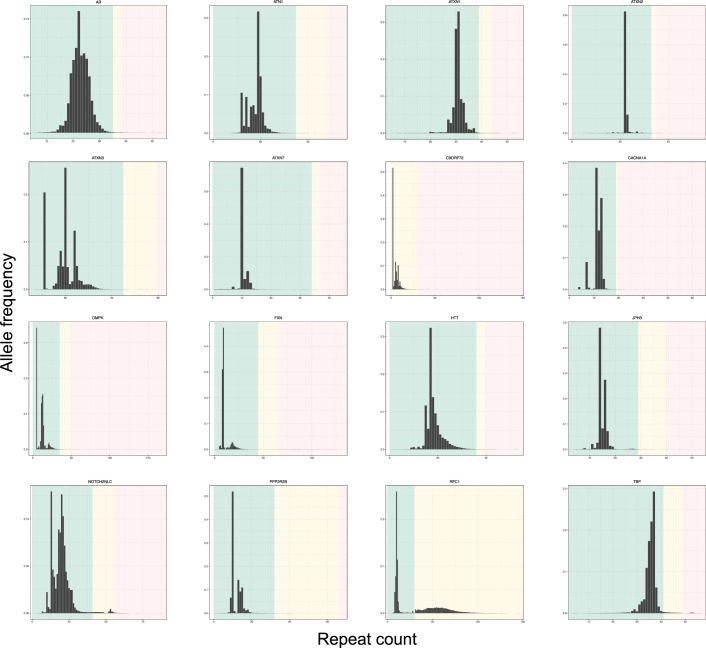
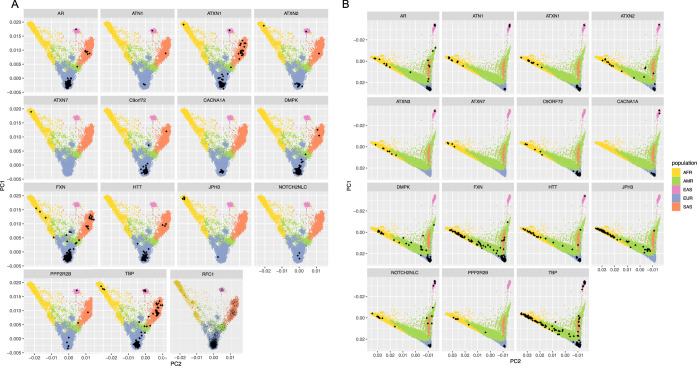
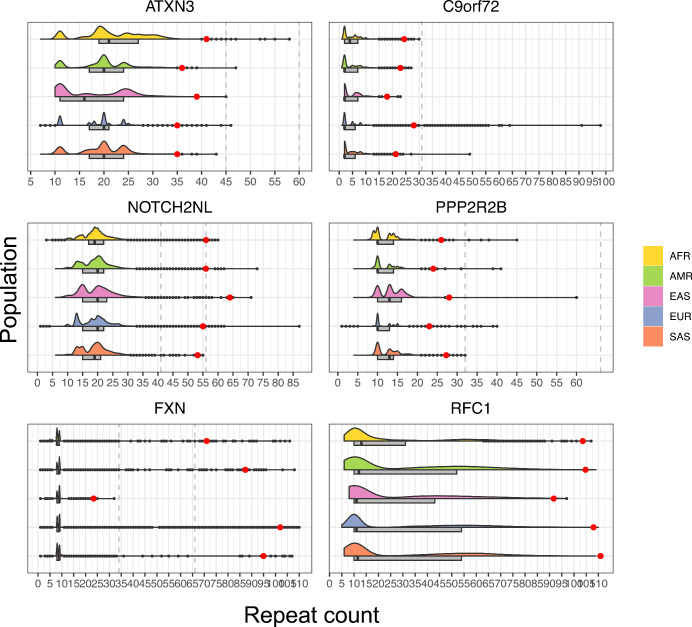
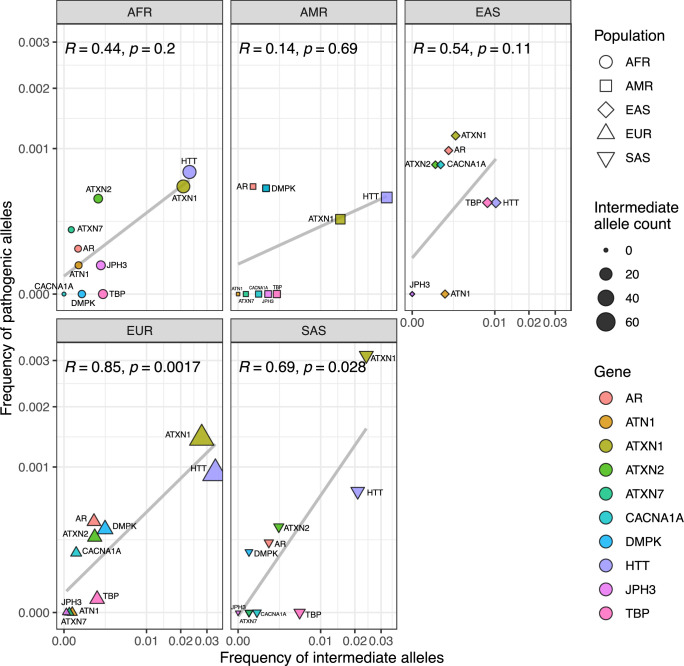
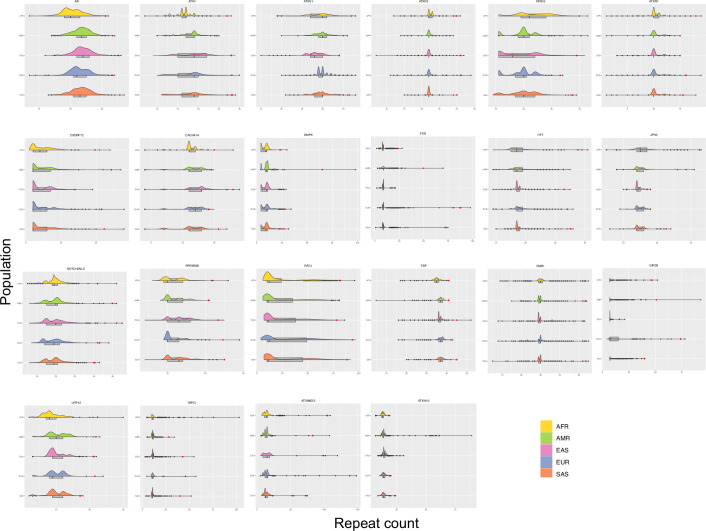
Repeat expansion disorders (REDs) are a devastating group of predominantly neurological diseases. Together they are common, affecting 1 in 3,000 people worldwide with population-specific differences. However, prevalence estimates of REDs are hampered by heterogeneous clinical presentation, variable geographic distributions and technological limitations leading to underascertainment. Here, leveraging whole-genome sequencing data from 82,176 individuals from different populations, we found an overall disease allele frequency of REDs of 1 in 283 individuals. Modeling disease prevalence using genetic data, age at onset and survival, we show that the expected number of people with REDs would be two to three times higher than currently reported figures, indicating underdiagnosis and/or incomplete penetrance. While some REDs are population specific, for example, Huntington disease-like 2 in Africans, most REDs are represented in all broad genetic ancestries (that is, Europeans, Africans, Americans, East Asians and South Asians), challenging the notion that some REDs are found only in specific populations. These results have worldwide implications for local and global health communities in the diagnosis and counseling of REDs.
© 2024. The Author(s).
Conflict of interest statement
Figures
Update of
-
Increased frequency of repeat expansion mutations across different populations.medRxiv [Preprint]. 2024 Jul 8:2023.07.03.23292162. doi: 10.1101/2023.07.03.23292162. medRxiv. 2024. Update in: Nat Med. 2024 Nov;30(11):3357-3368. doi: 10.1038/s41591-024-03190-5. PMID: 37461547 Free PMC article. Updated. Preprint.
References
-
- Gossye, H., Engelborghs, S., Van Broeckhoven, C. & van der Zee, J. C9orf72 Frontotemporal Dementia and/or Amyotrophic Lateral Sclerosis (Univ. Washington, 2020).
MeSH terms
Grants and funding
- HHSN268201600002C/HL/NHLBI NIH HHS/United States
- U01 HL080295/HL/NHLBI NIH HHS/United States
- U01 HL130114/HL/NHLBI NIH HHS/United States
- N01 HC095167/HL/NHLBI NIH HHS/United States
- R03 HD103782/HD/NICHD NIH HHS/United States
- U01 HL089856/HL/NHLBI NIH HHS/United States
- HHSN268201800015I/HB/NHLBI NIH HHS/United States
- HHSN268201700003I/HL/NHLBI NIH HHS/United States
- N01 HC085081/HL/NHLBI NIH HHS/United States
- N01 HC095160/HL/NHLBI NIH HHS/United States
- N01 HC085080/HL/NHLBI NIH HHS/United States
- HHSN268201500003C/HL/NHLBI NIH HHS/United States
- R01 NS105781/NS/NINDS NIH HHS/United States
- RF1 AG075051/AG/NIA NIH HHS/United States
- HHSN268201800012C/HL/NHLBI NIH HHS/United States
- N01 HC095161/HL/NHLBI NIH HHS/United States
- N01 HC095168/HL/NHLBI NIH HHS/United States
- R01 HL120393/HL/NHLBI NIH HHS/United States
- HHSN268201500001C/HL/NHLBI NIH HHS/United States
- UL1 TR001079/TR/NCATS NIH HHS/United States
- N01 HC095169/HL/NHLBI NIH HHS/United States
- N01 HC085082/HL/NHLBI NIH HHS/United States
- HHSN268201600018C/HL/NHLBI NIH HHS/United States
- HHSN268201800014I/HB/NHLBI NIH HHS/United States
- HHSN268200800007C/HL/NHLBI NIH HHS/United States
- N01 HC085086/HL/NHLBI NIH HHS/United States
- N01 HC085083/HL/NHLBI NIH HHS/United States
- U01 HL120393/HL/NHLBI NIH HHS/United States
- U01 HL089897/HL/NHLBI NIH HHS/United States
- HHSN268201800014C/HL/NHLBI NIH HHS/United States
- S10 OD018522/OD/NIH HHS/United States
- N01 HC095159/HL/NHLBI NIH HHS/United States
- P30 DK063491/DK/NIDDK NIH HHS/United States
- HHSN268201700002C/HL/NHLBI NIH HHS/United States
- HHSN268201200036C/HL/NHLBI NIH HHS/United States
- HHSN268201800001C/HL/NHLBI NIH HHS/United States
- HHSN268201700001I/HL/NHLBI NIH HHS/United States
- N01 HC025195/HL/NHLBI NIH HHS/United States
- UM1 HG008853/HG/NHGRI NIH HHS/United States
- HHSN268201800013I/MD/NIMHD NIH HHS/United States
- N01 HC055222/HL/NHLBI NIH HHS/United States
- HHSN268201600003C/HL/NHLBI NIH HHS/United States
- HHSN268201700004I/HL/NHLBI NIH HHS/United States
- HHSN268201500001I/HL/NHLBI NIH HHS/United States
- HHSN268201800012I/HL/NHLBI NIH HHS/United States
- U18 FD005320/FD/FDA HHS/United States
- N01 HC085079/HL/NHLBI NIH HHS/United States
- UL1 TR001420/TR/NCATS NIH HHS/United States
- HHSN268201600004C/HL/NHLBI NIH HHS/United States
- N01 HC095163/HL/NHLBI NIH HHS/United States
- HHSN268201800011C/HL/NHLBI NIH HHS/United States
- HHSN268201500014C/HL/NHLBI NIH HHS/United States
- HHSN268201500003I/HL/NHLBI NIH HHS/United States
- HHSN268201600001C/HL/NHLBI NIH HHS/United States
- S10 OD030463/OD/NIH HHS/United States
- HHSN268201700005C/HL/NHLBI NIH HHS/United States
- HHSN268201700001C/HL/NHLBI NIH HHS/United States
- HHSN268201700003C/HL/NHLBI NIH HHS/United States
- 75N92019D00031/HL/NHLBI NIH HHS/United States
- HHSN268201700004C/HL/NHLBI NIH HHS/United States
- UL1 TR000040/TR/NCATS NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- N01 HC095166/HL/NHLBI NIH HHS/United States
- HHSN268201700002I/HL/NHLBI NIH HHS/United States
- HHSN268201800010I/HB/NHLBI NIH HHS/United States
- HHSN268201700005I/HL/NHLBI NIH HHS/United States
- MR/S006753/1/RCUK | Medical Research Council (MRC)
- R01 AG023629/AG/NIA NIH HHS/United States
- R01 HL117626/HL/NHLBI NIH HHS/United States
- N01 HC095162/HL/NHLBI NIH HHS/United States
- HHSN268201800011I/HB/NHLBI NIH HHS/United States
- N01 HC095165/HL/NHLBI NIH HHS/United States
- N01 HC095164/HL/NHLBI NIH HHS/United States
- UL1 TR001881/TR/NCATS NIH HHS/United States
- U01 NS120241/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
