

A novel peptide-based tau aggregation inhibitor as a potential therapeutic for Alzheimer's disease and other tauopathies
- PMID: 39360630
- PMCID: PMC11567856
- DOI: 10.1002/alz.14246
A novel peptide-based tau aggregation inhibitor as a potential therapeutic for Alzheimer's disease and other tauopathies
Abstract
Introduction: As aggregation underpins Tau toxicity, aggregation inhibitor peptides may have disease-modifying potential. They are therefore currently being designed and target either the 306VQIVYK311 aggregation-promoting hotspot found in all Tau isoforms or the 275VQIINK280 aggregation-promoting hotspot found in 4R isoforms. However, for any Tau aggregation inhibitor to potentially be clinically relevant for other tauopathies, it should target both hotspots to suppress aggregation of Tau isoforms, be stable, cross the blood-brain barrier, and rescue aggregation-dependent Tau phenotypes in vivo.
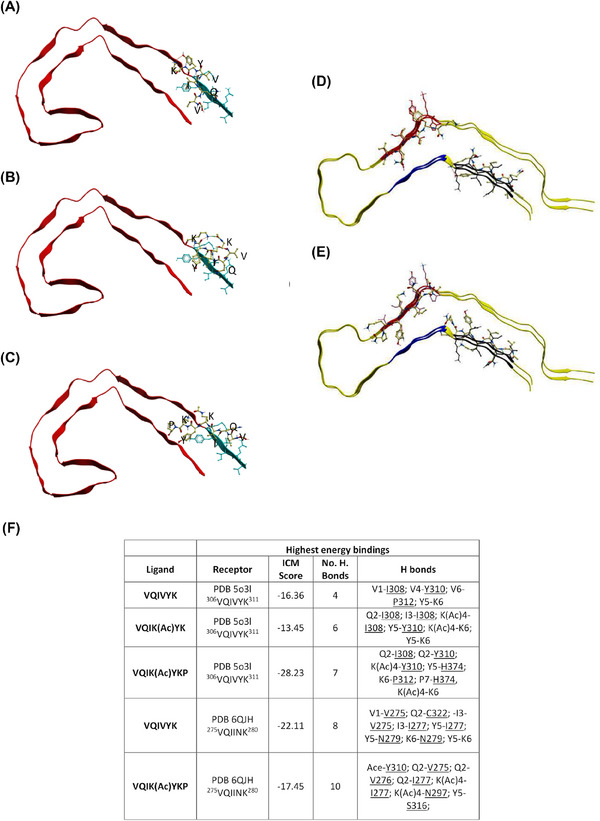
Methods: We developed a retro-inverso, stable D-amino peptide, RI-AG03 [Ac-rrrrrrrrGpkyk(ac)iqvGr-NH2], based on the 306VQIVYK311 hotspots which exhibit these disease-relevant attributes.
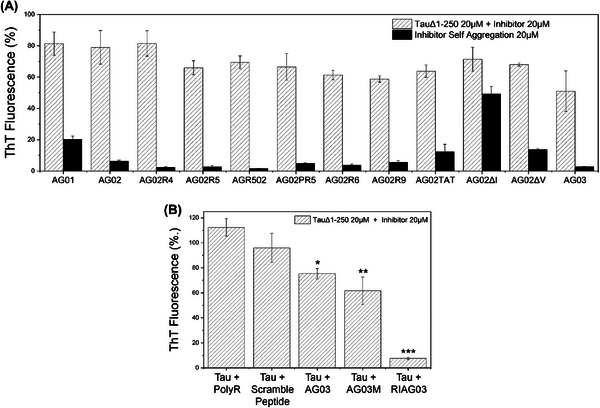
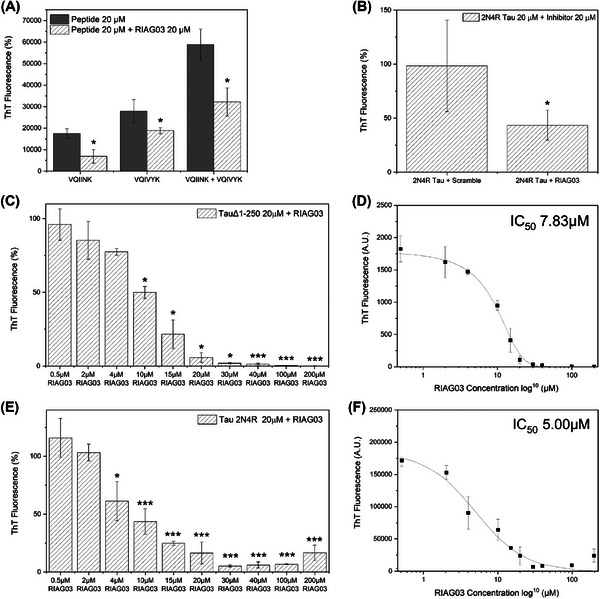
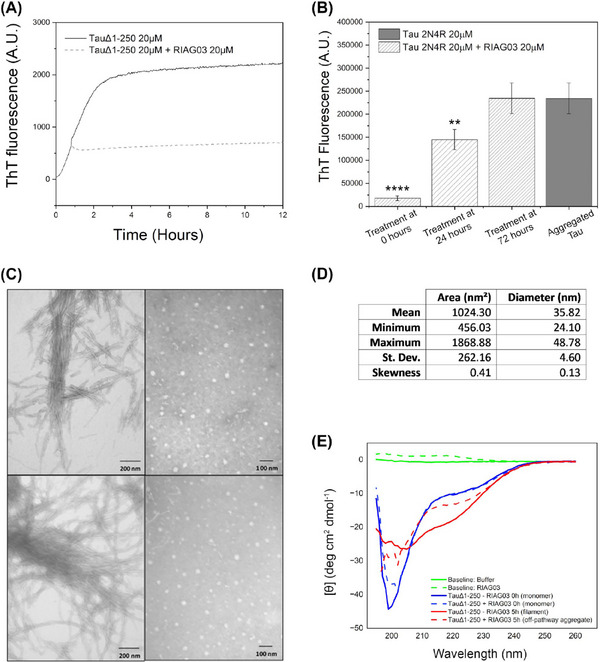
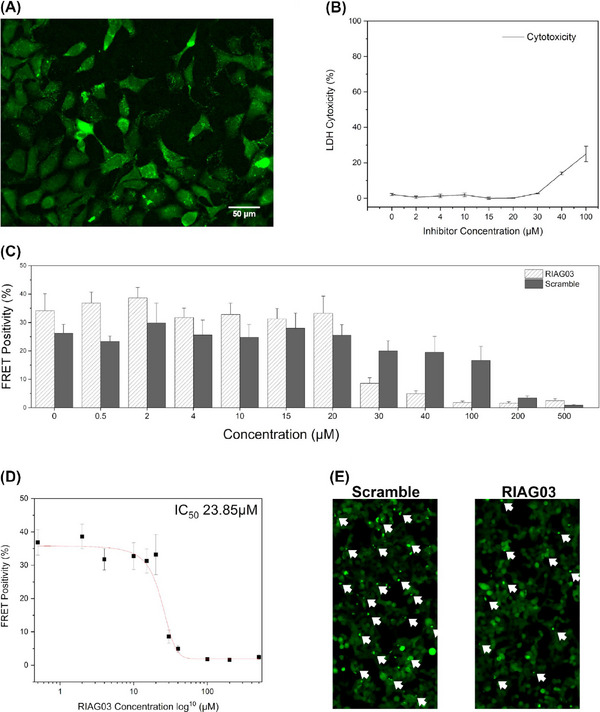
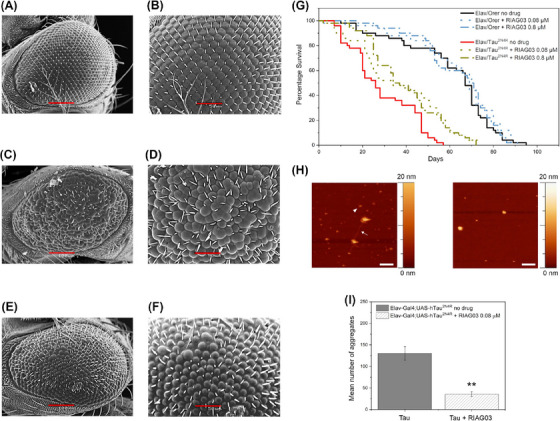
Results: Unlike other aggregation inhibitors, RI-AG03 effectively suppresses aggregation of multiple Tau species containing both hotspots in vitro and in vivo, is non-toxic, and suppresses aggregation-dependent neurodegenerative and behavioral phenotypes.
Discussion: RI-AG03 therefore meets many clinically relevant requirements for an anti-aggregation Tau therapeutic and should be explored further for its disease-modifying potential for Tauopathies.
Highlights: Our manuscript describes the development of a novel peptide inhibitor of Tau aggregation, a retro-inverso, stable D-amino peptide called RI-AG03 that displays many clinically relevant attributes. We show its efficacy in preventing Tau aggregation in both in vitro and in vivo experimental models while being non-toxic to cells. RI-AG03 also rescues a biosensor cell line that stably expresses Tau repeat domains with the P301S mutation fused to Cer/Clo and rescues aggregation-dependent phenotypes in vivo, suppressing neurodegeneration and extending lifespan. Collectively our data describe several properties and attributes of RI-AG03 that make it a promising disease-modifying candidate to explore for reducing pathogenic Tau aggregation in Tauopathies such as Alzheimer's disease. Given the real interest in reducing Tau aggregation and the potential clinical benefit of using such agents in clinical practice, RI-AG03 should be investigated further for the treatment of Tauopathies after validation in mammalian models. Tau aggregation inhibitors are the obvious first choice as Tau-based therapies as much of Tau-mediated toxicity is aggregation dependent. Indeed, there are many research efforts focusing on this therapeutic strategy with aggregation inhibitors being designed against one of the two aggregation-promoting hotspots of the Tau protein. To our knowledge, RI-AG03 is the only peptide aggregation inhibitor that inhibits aggregation of Tau by targeting both aggregation-promoting hotspot motifs simultaneously. As such, we believe that our study will have a significant impact on drug discovery efforts in this arena.
Keywords: Alzheimer's disease; Drosophila; VQIINK; VQIVYK; aggregation; amino‐acid; dementia; drug development; in silico; in vitro; in vivo; inhibitor; melanogaster; peptide; tau; tauopathies; therapeutic.
© 2024 The Author(s). Alzheimer's & Dementia published by Wiley Periodicals LLC on behalf of Alzheimer's Association.
Conflict of interest statement
The authors declare that they have no conflict of interest. Author disclosures are available in the supporting information.
Figures
References
-
- Mayes J, Tinker‐Mill C, Kolosov O, Zhang H, Tabner BJ, Allsop D. β‐amyloid fibrils in Alzheimer disease are not inert when bound to copper ions but can degrade hydrogen peroxide and generate reactive oxygen species. J Biol Chem. 2014;289(17):12052‐12062. doi:10.1074/jbc.M113.525212 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
