

Whole-genome sequencing in 333,100 individuals reveals rare non-coding single variant and aggregate associations with height
- PMID: 39362880
- PMCID: PMC11450065
- DOI: 10.1038/s41467-024-52579-w
Whole-genome sequencing in 333,100 individuals reveals rare non-coding single variant and aggregate associations with height
Abstract
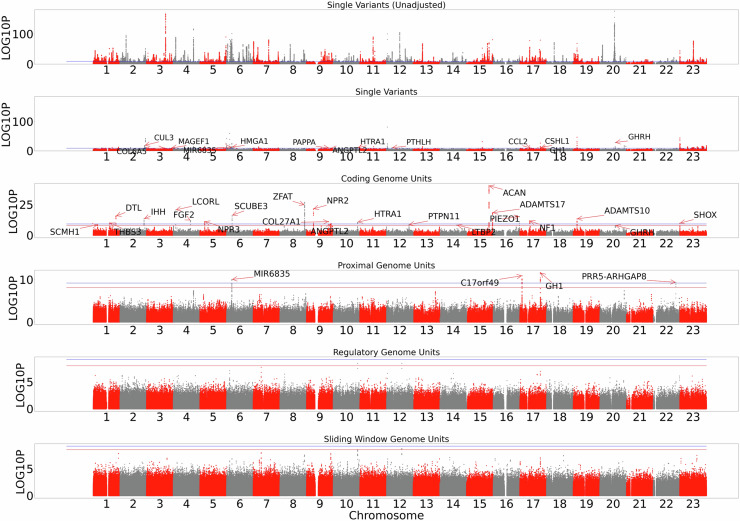
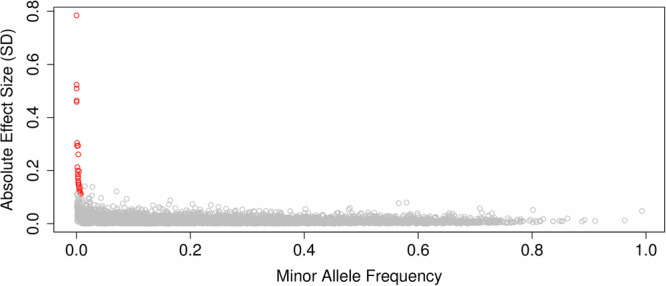
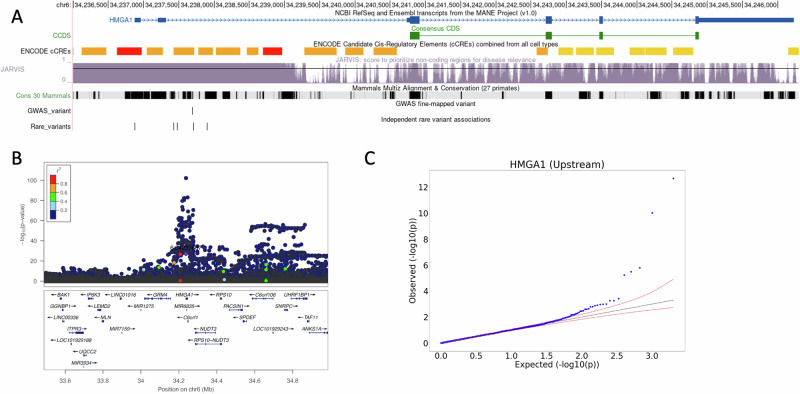
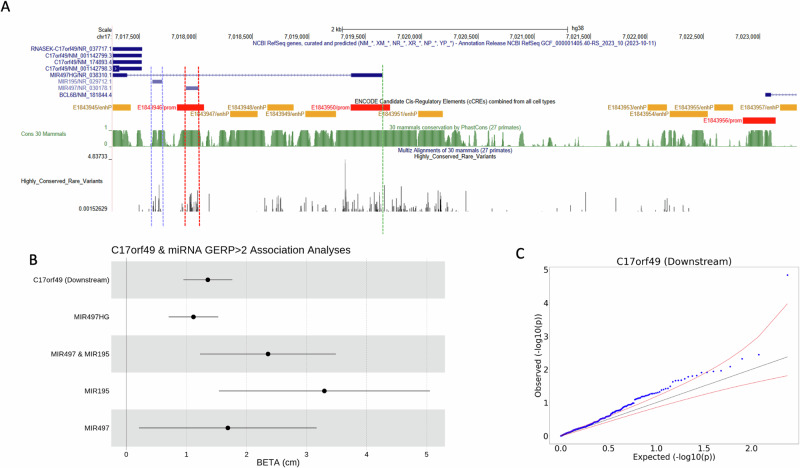
The role of rare non-coding variation in complex human phenotypes is still largely unknown. To elucidate the impact of rare variants in regulatory elements, we performed a whole-genome sequencing association analysis for height using 333,100 individuals from three datasets: UK Biobank (N = 200,003), TOPMed (N = 87,652) and All of Us (N = 45,445). We performed rare ( < 0.1% minor-allele-frequency) single-variant and aggregate testing of non-coding variants in regulatory regions based on proximal-regulatory, intergenic-regulatory and deep-intronic annotation. We observed 29 independent variants associated with height at P < after conditioning on previously reported variants, with effect sizes ranging from -7cm to +4.7 cm. We also identified and replicated non-coding aggregate-based associations proximal to HMGA1 containing variants associated with a 5 cm taller height and of highly-conserved variants in MIR497HG on chromosome 17. We have developed an approach for identifying non-coding rare variants in regulatory regions with large effects from whole-genome sequencing data associated with complex traits.
© 2024. The Author(s).
Conflict of interest statement
Bruce M. Psaty serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. Xihong Lin is a consultant of AbbVie Pharmaceuticals and Verily Life Sciences. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
