

Population-specific putative causal variants shape quantitative traits
- PMID: 39363016
- PMCID: PMC11525193
- DOI: 10.1038/s41588-024-01913-5
Population-specific putative causal variants shape quantitative traits
Abstract
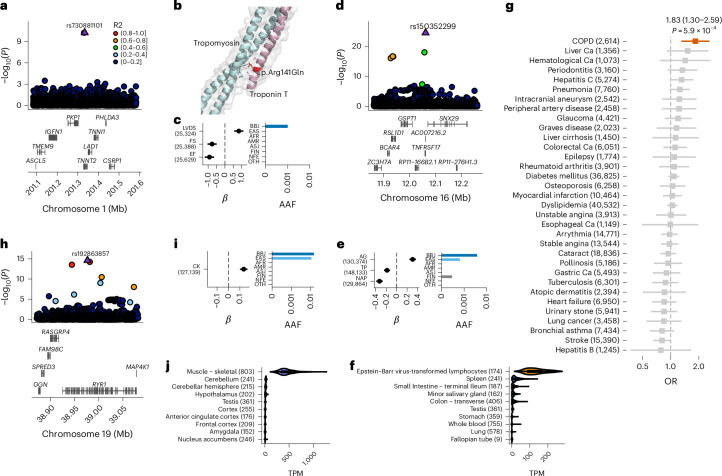
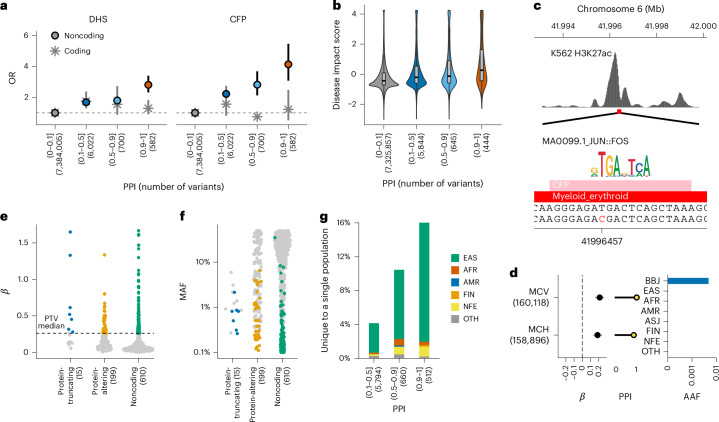
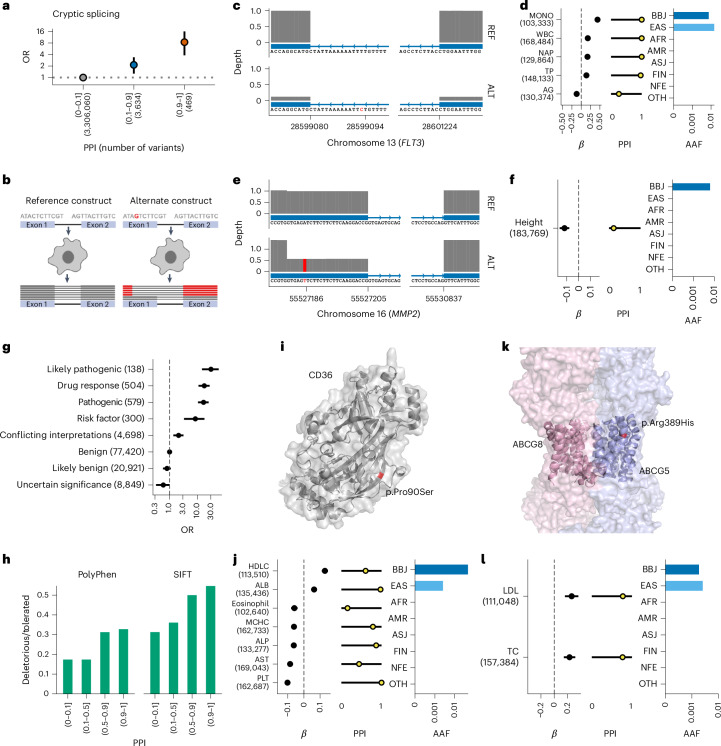
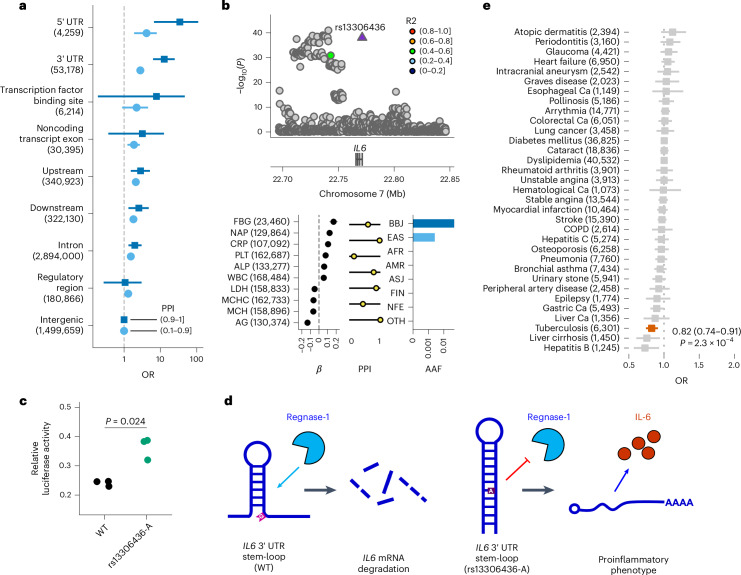
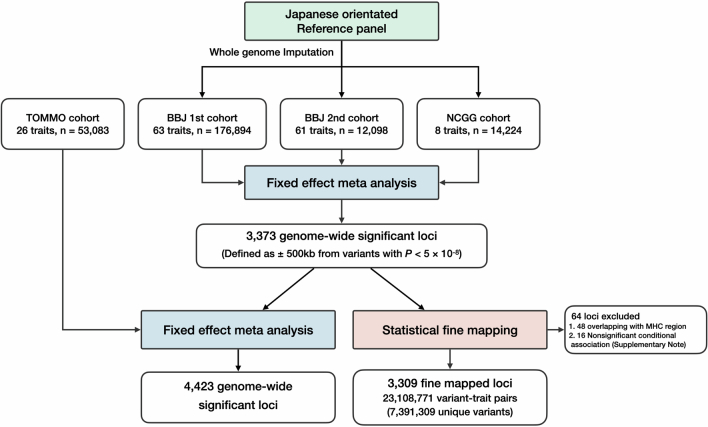
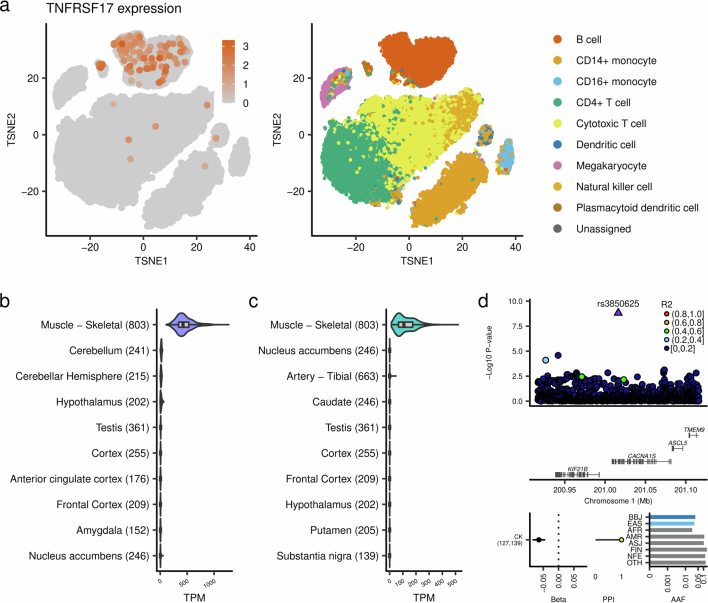
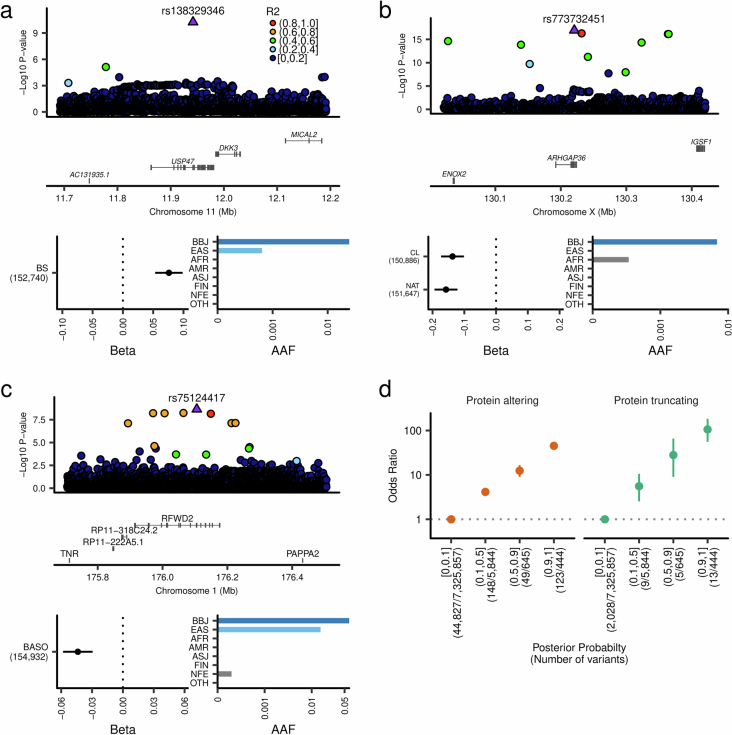
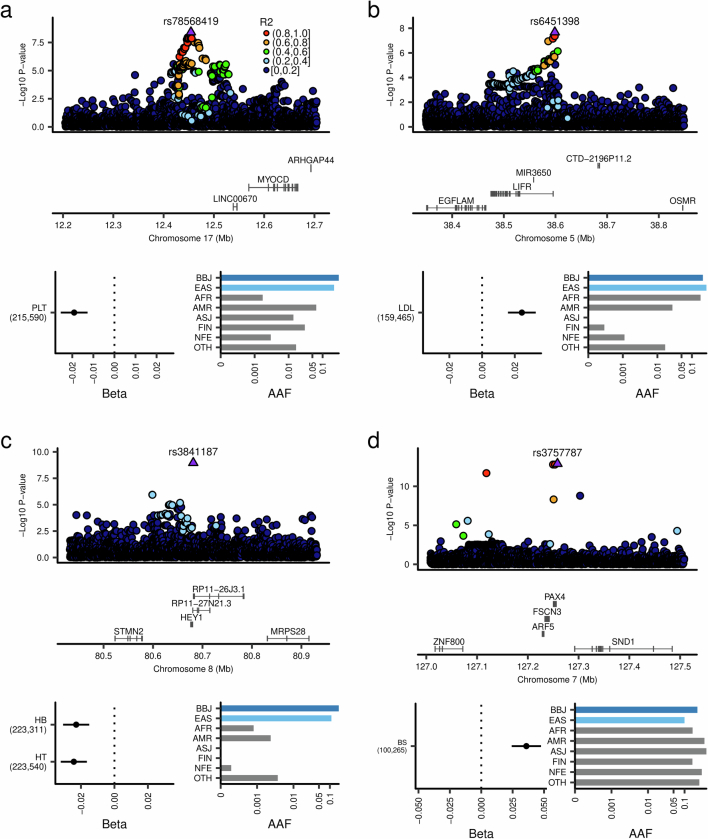
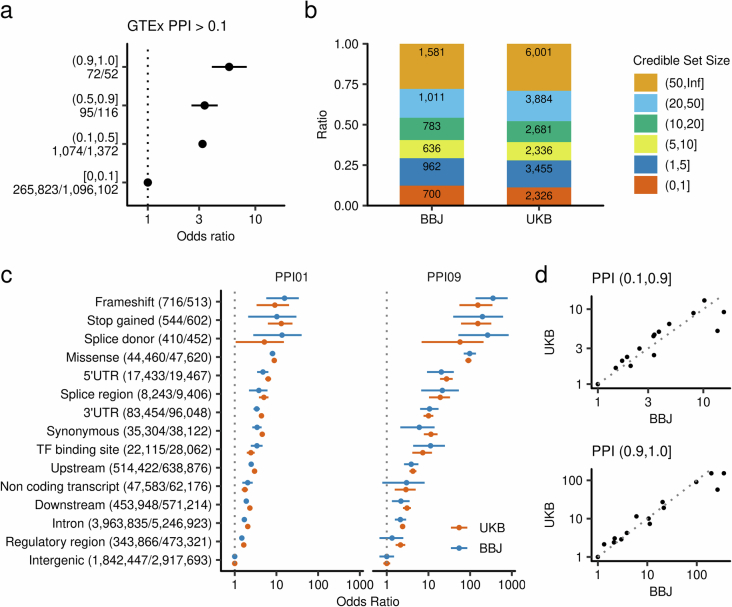
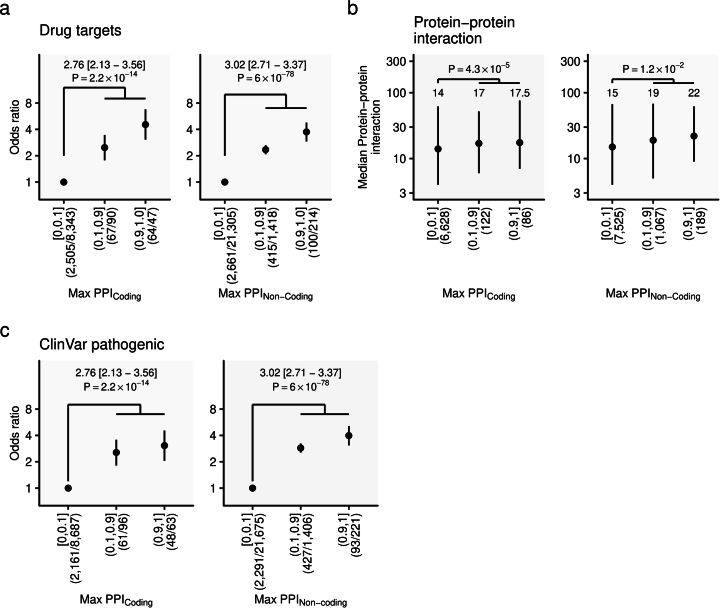
Human genetic variants are associated with many traits through largely unknown mechanisms. Here, combining approximately 260,000 Japanese study participants, a Japanese-specific genotype reference panel and statistical fine-mapping, we identified 4,423 significant loci across 63 quantitative traits, among which 601 were new, and 9,406 putatively causal variants. New associations included Japanese-specific coding, splicing and noncoding variants, exemplified by a damaging missense variant rs730881101 in TNNT2 associated with lower heart function and increased risk for heart failure (P = 1.4 × 10-15 and odds ratio = 4.5, 95% confidence interval = 3.1-6.5). Putative causal noncoding variants were supported by state-of-art in silico functional assays and had comparable effect sizes to coding variants. A plausible example of new mechanisms of causal variants is an enrichment of causal variants in 3' untranslated regions (UTRs), including the Japanese-specific rs13306436 in IL6 associated with pro-inflammatory traits and protection against tuberculosis. We experimentally showed that transcripts with rs13306436 are resistant to mRNA degradation by regnase-1, an RNA-binding protein. Our study provides a list of fine-mapped causal variants to be tested for functionality and underscores the importance of sequencing, genotyping and association efforts in diverse populations.
© 2024. The Author(s).
Conflict of interest statement
P.N. reports research grants from Allelica, Amgen, Apple, Boston Scientific, Genentech/Roche and Novartis; personal fees from Allelica, Apple, AstraZeneca, Blackstone Life Sciences, Creative Education Concepts, CRISPR Therapeutics, Eli Lilly & Co, Foresite Labs, Genentech/Roche, GV, HeartFlow, Magnet Biomedicine, Merck and Novartis; and scientific advisory board membership of Esperion Therapeutics, Preciseli and TenSixteen Bio. He is scientific cofounder of TenSixteen Bio; holds equity in MyOme, Preciseli and TenSixteen Bio; and reports spousal employment at Vertex Pharmaceuticals, all unrelated to the present work. The other authors declare no competing interests.
Figures
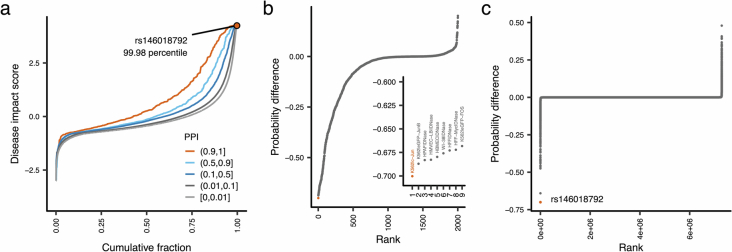
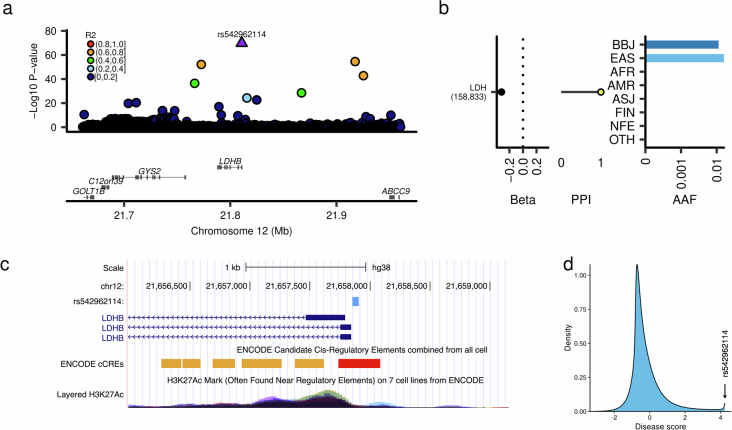
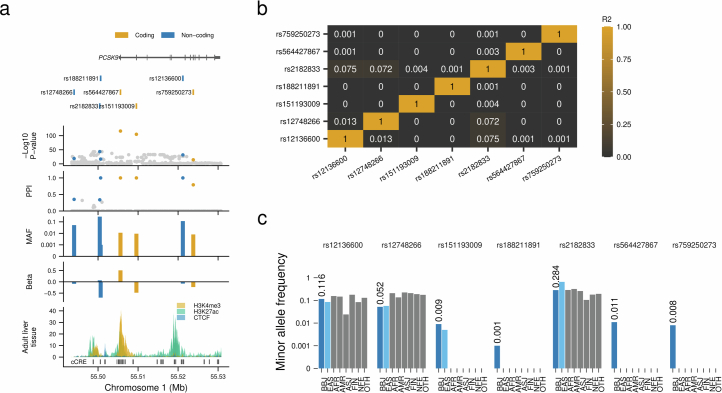
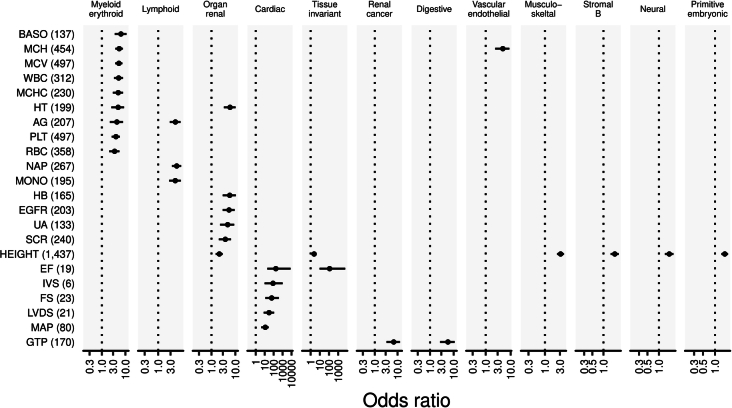
References
-
- Akiyama, M. et al. Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat. Genet.49, 1458–1467 (2017). - PubMed
MeSH terms
Substances
Grants and funding
- 21kk0305013/Japan Agency for Medical Research and Development (AMED)
- 21tm0424220/Japan Agency for Medical Research and Development (AMED)
- 21ck0106642/Japan Agency for Medical Research and Development (AMED)
- 18km0605001/Japan Agency for Medical Research and Development (AMED)
- 21ae0121030/Japan Agency for Medical Research and Development (AMED)
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
