

De novo GTP synthesis is a metabolic vulnerability for the interception of brain metastases
- PMID: 39366383
- PMCID: PMC11513854
- DOI: 10.1016/j.xcrm.2024.101755
De novo GTP synthesis is a metabolic vulnerability for the interception of brain metastases
Abstract
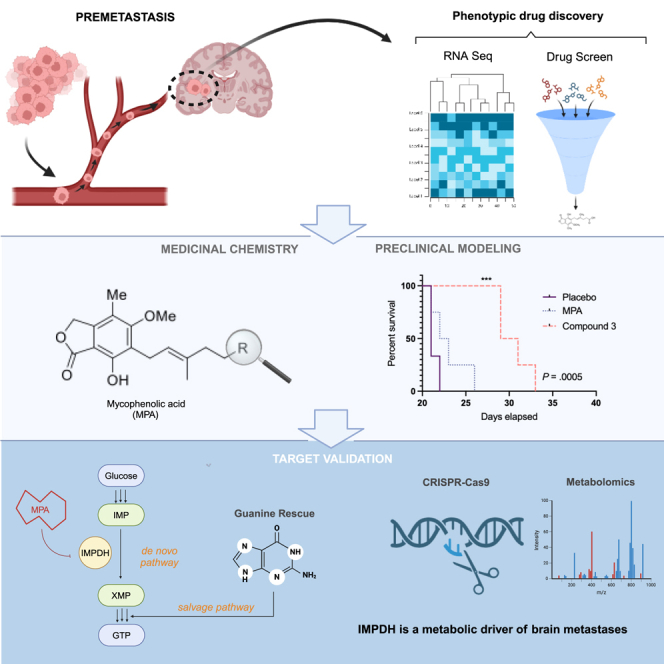
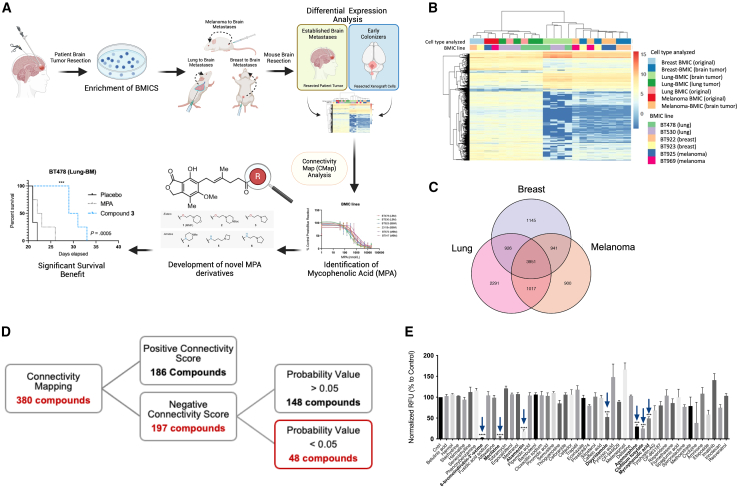
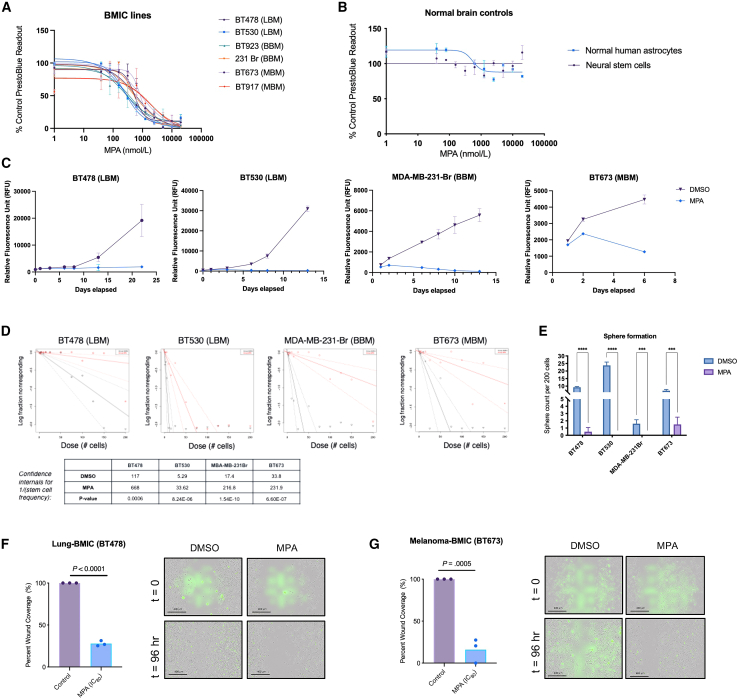
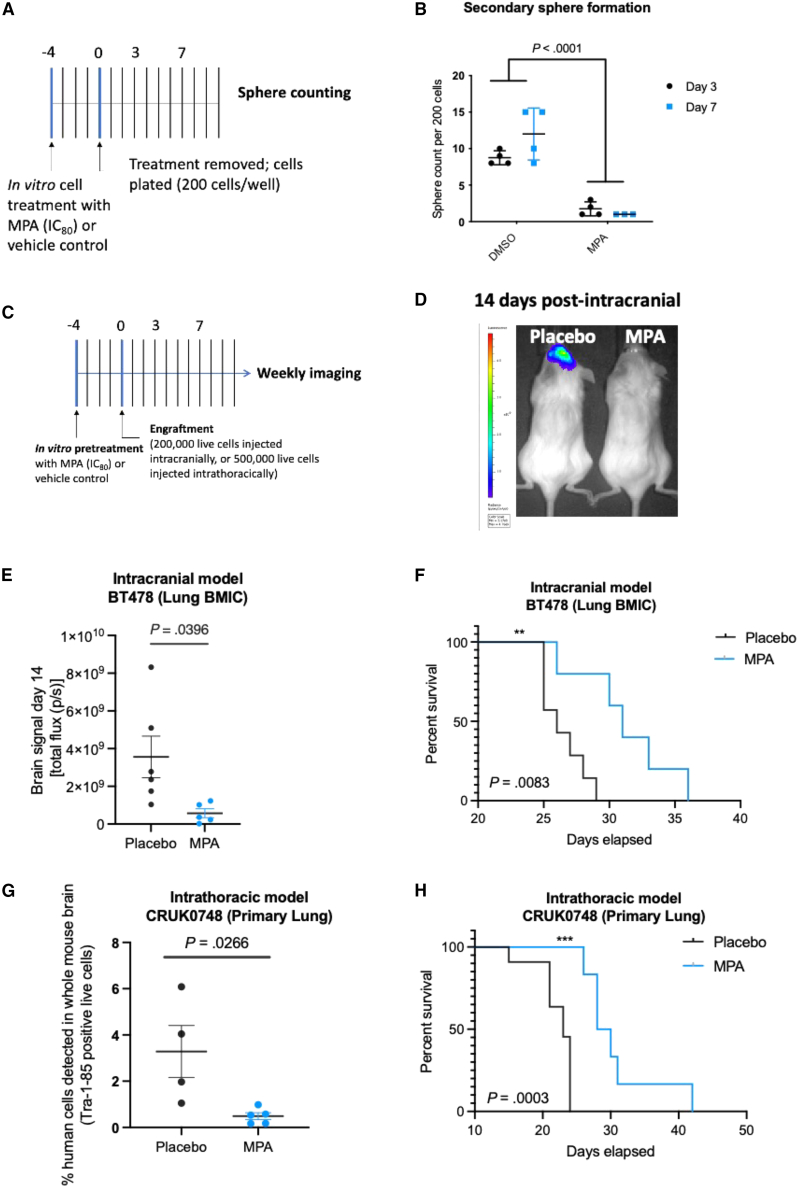
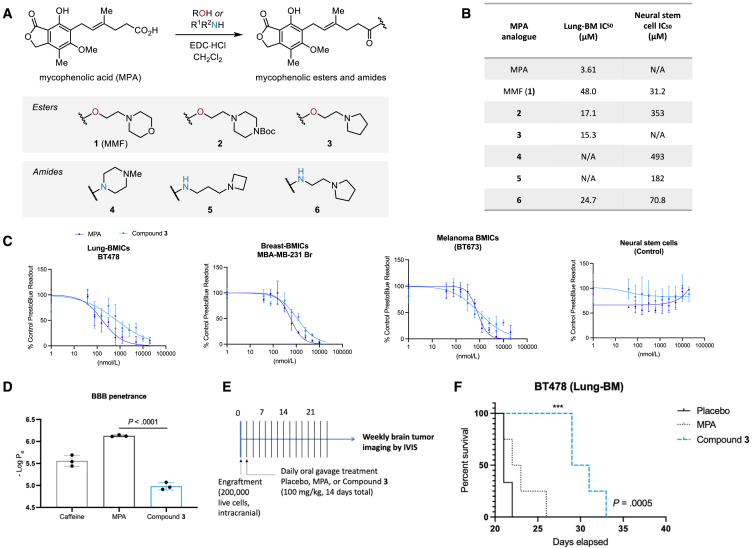
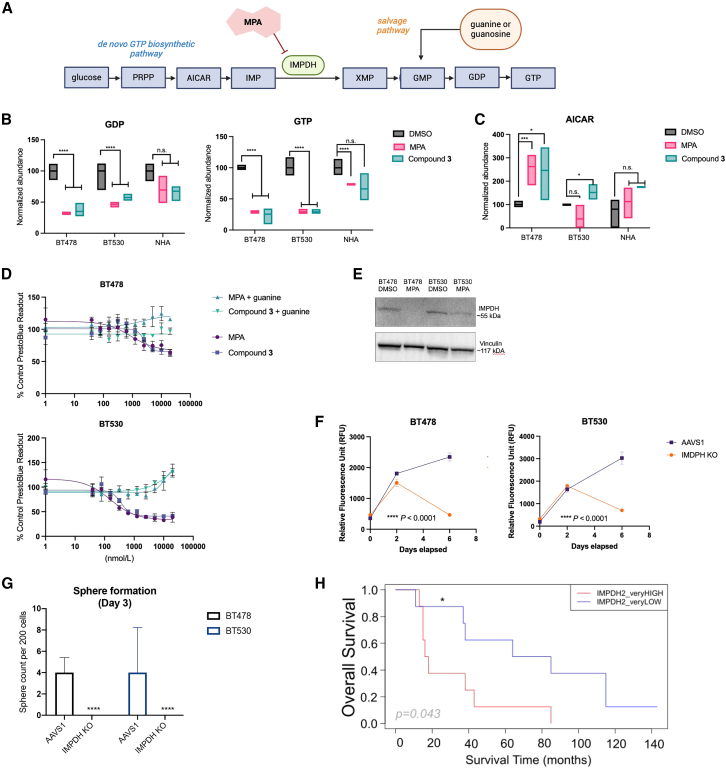
Patients with brain metastases (BM) face a 90% mortality rate within one year of diagnosis and the current standard of care is palliative. Targeting BM-initiating cells (BMICs) is a feasible strategy to treat BM, but druggable targets are limited. Here, we apply Connectivity Map analysis to lung-, breast-, and melanoma-pre-metastatic BMIC gene expression signatures and identify inosine monophosphate dehydrogenase (IMPDH), the rate-limiting enzyme in the de novo GTP synthesis pathway, as a target for BM. We show that pharmacological and genetic perturbation of IMPDH attenuates BMIC proliferation in vitro and the formation of BM in vivo. Metabolomic analyses and CRISPR knockout studies confirm that de novo GTP synthesis is a potent metabolic vulnerability in BM. Overall, our work employs a phenotype-guided therapeutic strategy to uncover IMPDH as a relevant target for attenuating BM outgrowth, which may provide an alternative treatment strategy for patients who are otherwise limited to palliation.
Keywords: GTP synthesis; IMPDH; brain metastases; cancer stem cells.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A.M.K., J.W.J., C.V., J.M., and S.K.S. are listed as co-inventors for a PCT patent that has been filed, relating to this work.
Figures
References
-
- Soffietti R., Cornu P., Delattre J.Y. 2011. Brain metastases 10.1038/s41572-018-0055-y.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
