

Reversible covalent c-Jun N-terminal kinase inhibitors targeting a specific cysteine by precision-guided Michael-acceptor warheads
- PMID: 39366946
- PMCID: PMC11452492
- DOI: 10.1038/s41467-024-52573-2
Reversible covalent c-Jun N-terminal kinase inhibitors targeting a specific cysteine by precision-guided Michael-acceptor warheads
Abstract
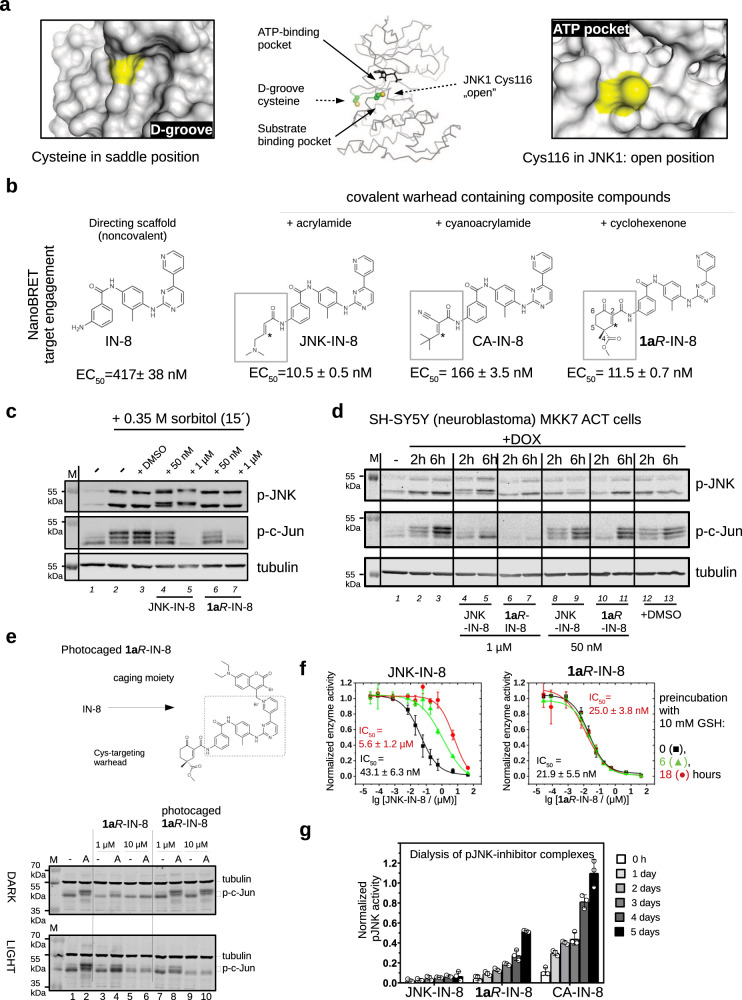
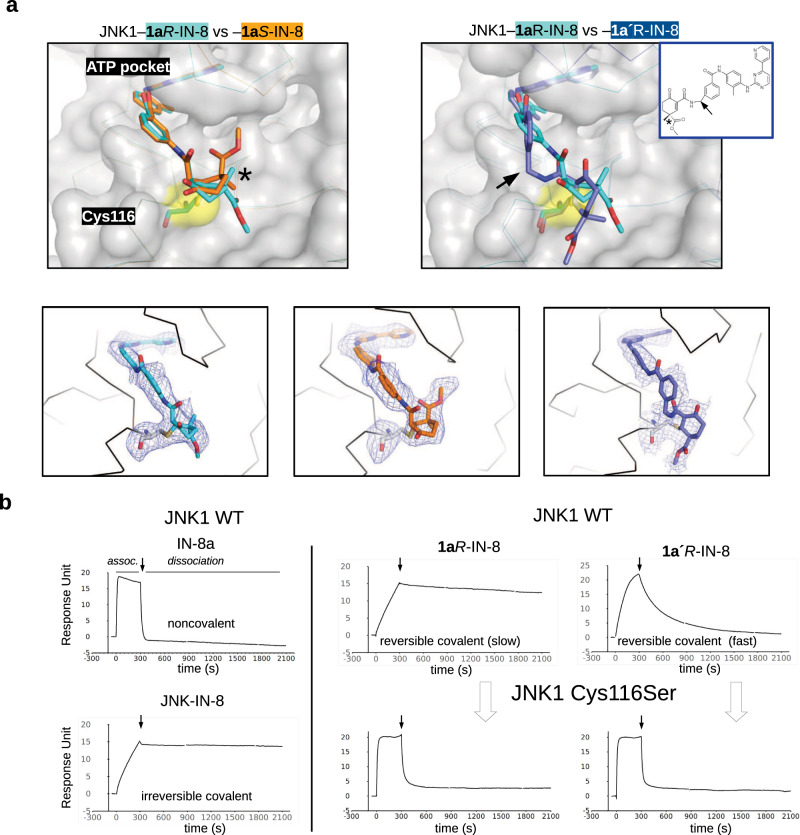
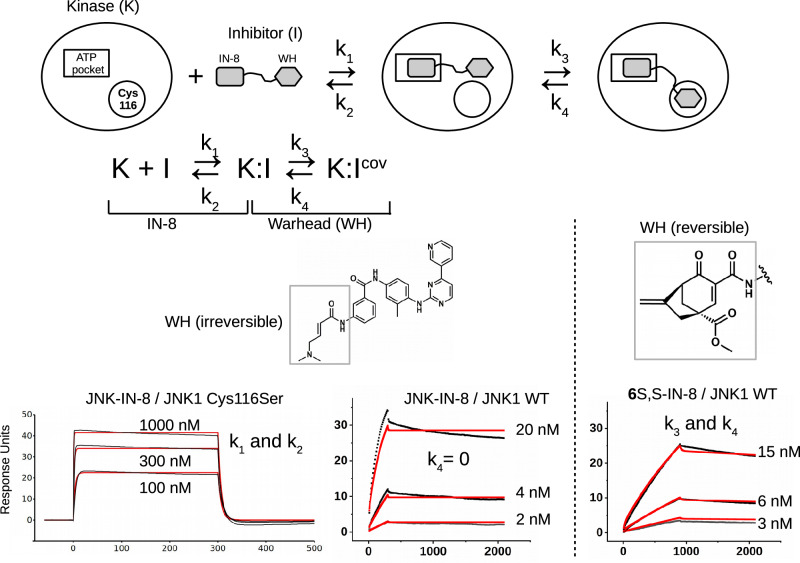
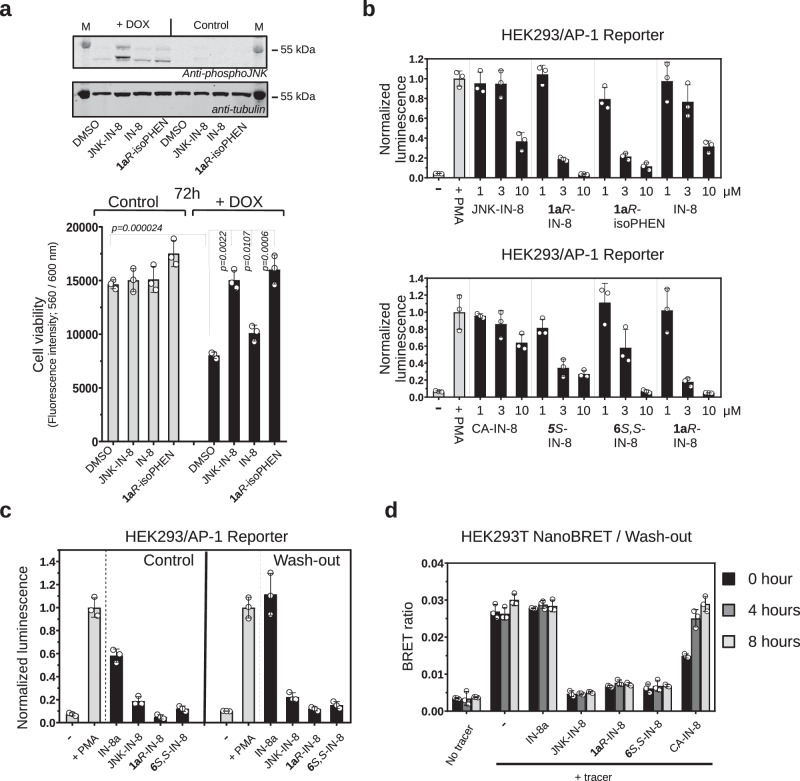
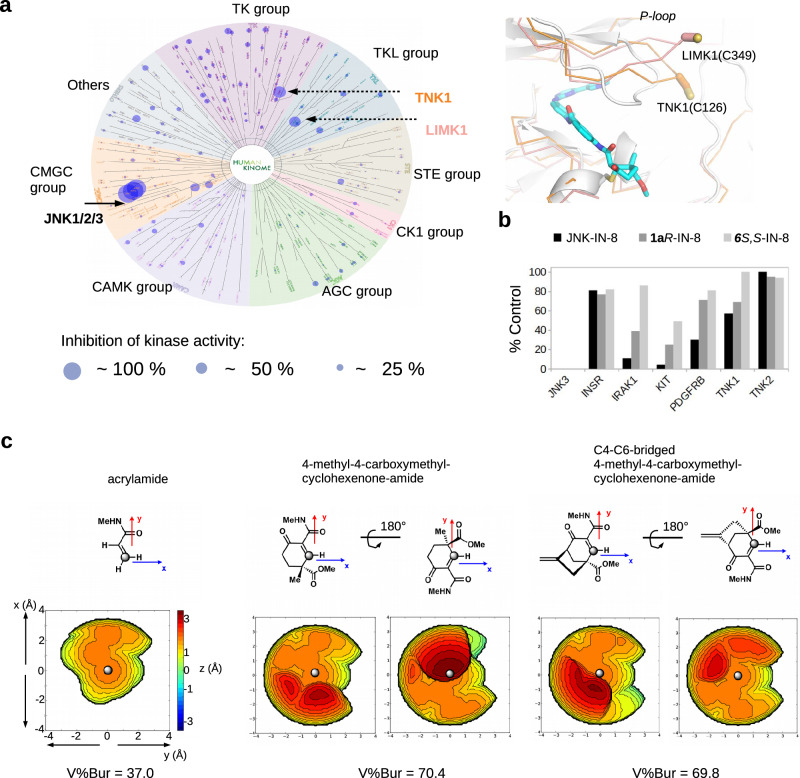
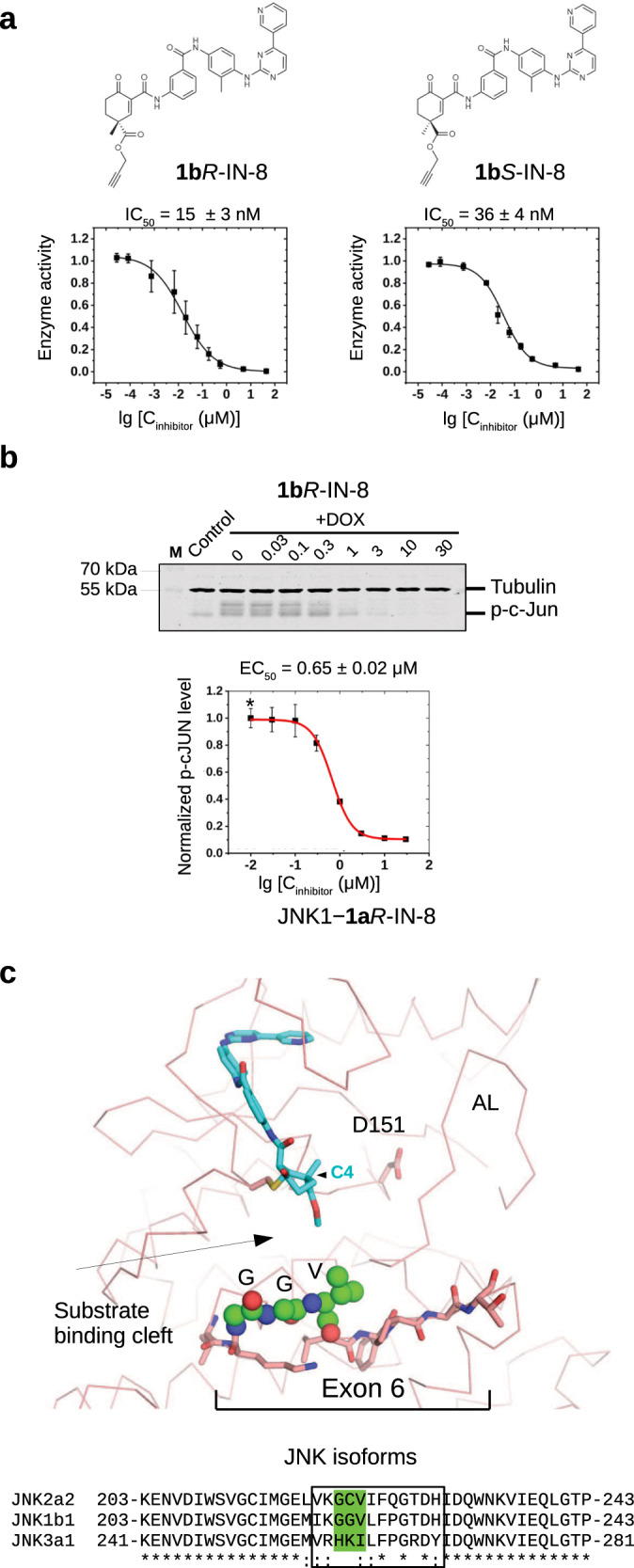
There has been a surge of interest in covalent inhibitors for protein kinases in recent years. Despite success in oncology, the off-target reactivity of these molecules is still hampering the use of covalent warhead-based strategies. Herein, we disclose the development of precision-guided warheads to mitigate the off-target challenge. These reversible warheads have a complex and cyclic structure with optional chirality center and tailored steric and electronic properties. To validate our proof-of-concept, we modified acrylamide-based covalent inhibitors of c-Jun N-terminal kinases (JNKs). We show that the cyclic warheads have high resilience against off-target thiols. Additionally, the binding affinity, residence time, and even JNK isoform specificity can be fine-tuned by adjusting the substitution pattern or using divergent and orthogonal synthetic elaboration of the warhead. Taken together, the cyclic warheads presented in this study will be a useful tool for medicinal chemists for the deliberate design of safer and functionally fine-tuned covalent inhibitors.
© 2024. The Author(s).
Conflict of interest statement
A.R., T.S., Á.L.P., D.B., A.A., P.S., L.T., K.A., T.I., E.SZ. and R.P. are inventors in a patent application pending approval (PCT/HU2023/050079) on the use of cyclic designer scaffolds for the covalent targeting of proteins via Michael addition. The remaining authors declare no competing interests.
Figures
Similar articles
-
Targeting a key protein-protein interaction surface on mitogen-activated protein kinases by a precision-guided warhead scaffold.Nat Commun. 2024 Oct 4;15(1):8607. doi: 10.1038/s41467-024-52574-1. Nat Commun. 2024. PMID: 39366929 Free PMC article.
-
c-Jun N-terminal kinase inhibitors: a patent review (2010 - 2014).Expert Opin Ther Pat. 2015;25(8):849-72. doi: 10.1517/13543776.2015.1039984. Epub 2015 May 19. Expert Opin Ther Pat. 2015. PMID: 25991433 Review.
-
Exploring Extended Warheads toward Developing Cysteine-Targeted Covalent Kinase Inhibitors.J Chem Inf Model. 2024 Dec 23;64(24):9517-9527. doi: 10.1021/acs.jcim.4c00890. Epub 2024 Dec 10. J Chem Inf Model. 2024. PMID: 39656065 Free PMC article.
-
Inhibitors of c-Jun N-terminal kinases: an update.J Med Chem. 2015 Jan 8;58(1):72-95. doi: 10.1021/jm501212r. Epub 2014 Dec 8. J Med Chem. 2015. PMID: 25415535 Review.
-
Live-cell imaging and profiling of c-Jun N-terminal kinases using covalent inhibitor-derived probes.Chem Commun (Camb). 2019 Jan 25;55(8):1092-1095. doi: 10.1039/c8cc09558b. Epub 2019 Jan 8. Chem Commun (Camb). 2019. PMID: 30620026
Cited by
-
Covalent Targeting Leads to the Development of a LIMK1 Isoform-Selective Inhibitor.J Med Chem. 2025 Jul 24;68(14):15026-15049. doi: 10.1021/acs.jmedchem.5c01204. Epub 2025 Jul 2. J Med Chem. 2025. PMID: 40598933 Free PMC article.
-
Advancing Covalent Ligand and Drug Discovery beyond Cysteine.Chem Rev. 2025 Jul 23;125(14):6653-6684. doi: 10.1021/acs.chemrev.5c00001. Epub 2025 May 22. Chem Rev. 2025. PMID: 40404146 Free PMC article. Review.
-
Key advances in the development of reversible covalent inhibitors.Future Med Chem. 2025 Feb;17(4):389-392. doi: 10.1080/17568919.2025.2453407. Epub 2025 Jan 19. Future Med Chem. 2025. PMID: 39829174 No abstract available.
-
Methods for kinetic evaluation of reversible covalent inhibitors from time-dependent IC50 data.RSC Med Chem. 2025 Mar 20;16(6):2517-2531. doi: 10.1039/d5md00050e. eCollection 2025 Jun 18. RSC Med Chem. 2025. PMID: 40162199 Free PMC article.
-
Targeting a key protein-protein interaction surface on mitogen-activated protein kinases by a precision-guided warhead scaffold.Nat Commun. 2024 Oct 4;15(1):8607. doi: 10.1038/s41467-024-52574-1. Nat Commun. 2024. PMID: 39366929 Free PMC article.
References
-
- Roskoski, R. Orally effective FDA-approved protein kinase targeted covalent inhibitors (TCIs). Pharmacol. Res.165, 105422 (2021). - PubMed
-
- Gehringer, M. & Laufer, S. A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem.62, 5673–5724 (2019). - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
