

Whole-exome sequencing reveals the genetic causes and modifiers of moyamoya syndrome
- PMID: 39367156
- PMCID: PMC11452616
- DOI: 10.1038/s41598-024-72043-5
Whole-exome sequencing reveals the genetic causes and modifiers of moyamoya syndrome
Abstract
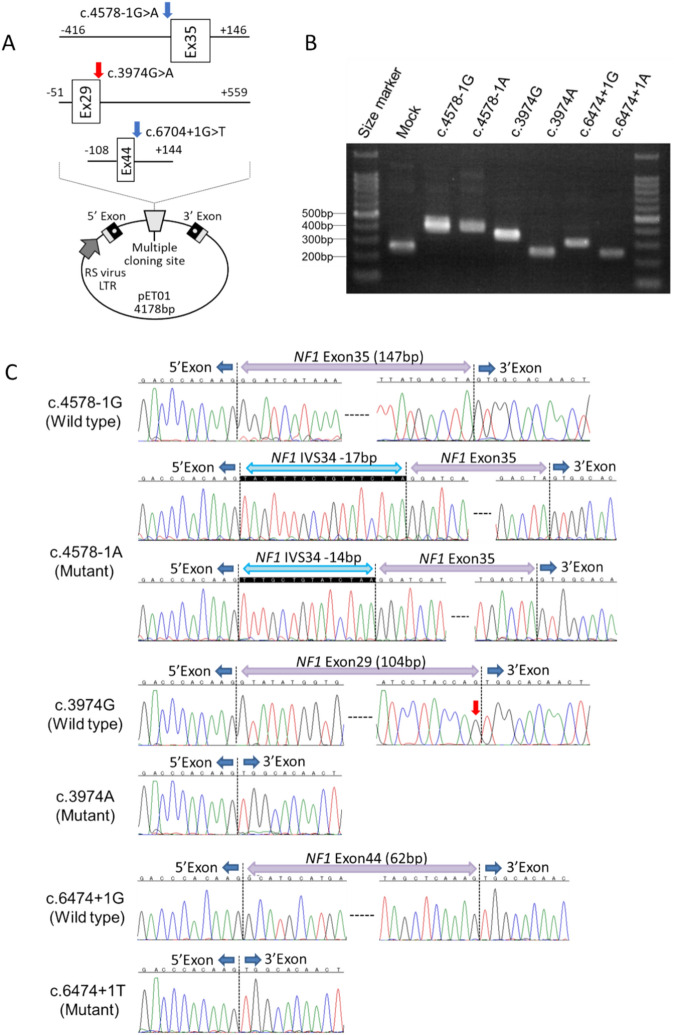
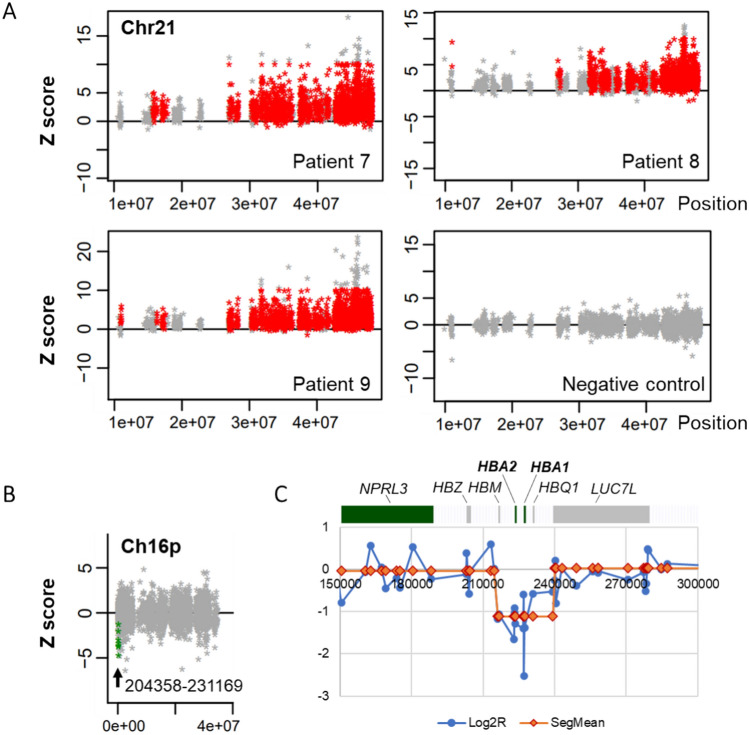
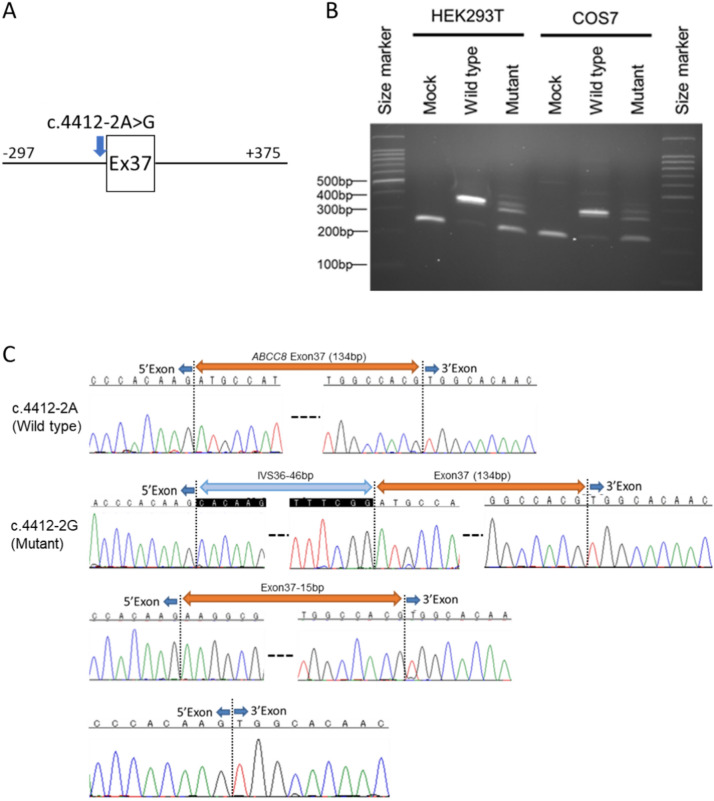
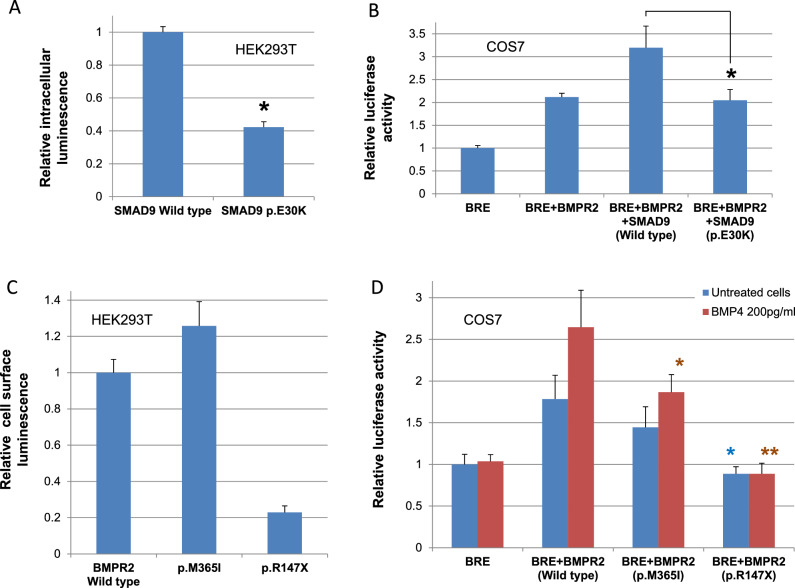
Moyamoya vasculopathy secondary to various genetic disorders is classified as moyamoya syndrome (MMS). Recent studies indicate MMS occurs due to a combination of genetic modifiers and causative mutations for the primary genetic disorders. We performed whole-exome sequencing (WES) in 13 patients with various genetic disorders who developed MMS. WES successfully revealed the genetic diagnoses of neurofibromatosis type 1 (NF-1), Down syndrome, multisystemic smooth muscle dysfunction syndrome, Noonan syndrome, and alpha thalassemia. The previously reported modifier genes, RNF213 and MRVI1, were confirmed in the NF-1 and Down syndrome cases. Further analysis revealed rare hypomorphic variants in the causative genes of the primary disorders underlying MMS, such as Alagille syndrome and Rasopathies, conferred susceptibility to MMS. Genes involved in the development of pulmonary arterial hypertension (PAH), such as ABCC8 and BMPR2, were also identified as potential modifiers. The rare variants in the MMS and PAH genes were significantly enriched in the eight Japanese patients with MMS compared with the 104 Japanese individuals from the 1000 Genomes Project. Disease genes associated with the arterial occlusive conditions represented by those of Rasopathies and PAH may provide novel diagnostic markers and future therapeutic targets for MMS as well as moyamoya disease with an unknown cause.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Research Committee on the Pathology and Treatment of Spontaneous Occlusion of the Circle of Willis, Health Labour Sciences Research Grant for Research on Measures for Infractable Diseases. Guidelines for diagnosis and treatment of moyamoya disease (spontaneous occlusion of the circle of Willis). Neurol. Med. Chir. (Tokyo)52(5), 245–266. 10.2176/nmc.52.245 (2012). - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
