

Leveraging explainable deep learning methodologies to elucidate the biological underpinnings of Huntington's disease using single-cell RNA sequencing data
- PMID: 39367331
- PMCID: PMC11451194
- DOI: 10.1186/s12864-024-10855-5
Leveraging explainable deep learning methodologies to elucidate the biological underpinnings of Huntington's disease using single-cell RNA sequencing data
Abstract
Background: Huntington's disease (HD) is a hereditary neurological disorder caused by mutations in HTT, leading to neuronal degeneration. Traditionally, HD is associated with the misfolding and aggregation of mutant huntingtin due to an extended polyglutamine domain encoded by an expanded CAG tract. However, recent research has also highlighted the role of global transcriptional dysregulation in HD pathology. However, understanding the intricate relationship between mRNA expression and HD at the cellular level remains challenging. Our study aimed to elucidate the underlying mechanisms of HD pathology using single-cell sequencing data.
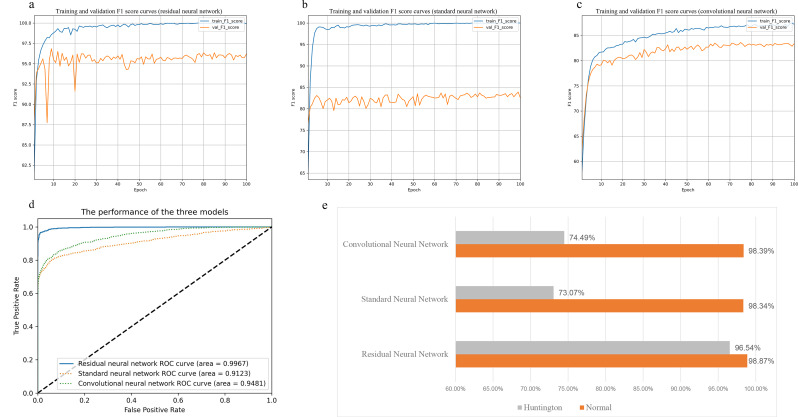
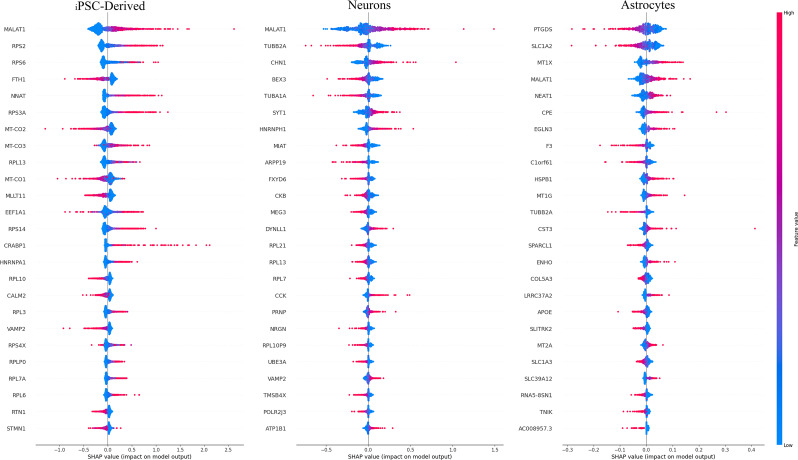
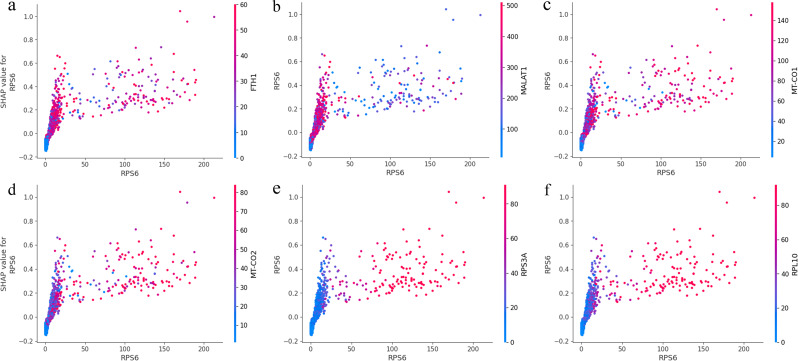
Results: We used single-cell RNA sequencing analysis to determine differential gene expression patterns between healthy and HD cells. HD cells were effectively modeled using a residual neural network (ResNet), which outperformed traditional and convolutional neural networks. Despite the efficacy of our approach, the F1 score for the test set was 96.53%. Using the SHapley Additive exPlanations (SHAP) algorithm, we identified genes influencing HD prediction and revealed their roles in HD pathobiology, such as in the regulation of cellular iron metabolism and mitochondrial function. SHAP analysis also revealed low-abundance genes that were overlooked by traditional differential expression analysis, emphasizing its effectiveness in identifying biologically relevant genes for distinguishing between healthy and HD cells. Overall, the integration of single-cell RNA sequencing data and deep learning models provides valuable insights into HD pathology.
Conclusion: We developed the model capable of analyzing HD at single-cell transcriptomic level.
Keywords: Deep residual model; Huntington’s disease; Single-cell RNA-seq; Unveiling potentially key genes.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- Sharma V, Sharma P, Deshmukh R. Huntington’s disease: clinical complexities and therapeutic strategies. J Adv Sci Res. 2012;3(02):30–6.
-
- Trepte P, Strempel N, Wanker EE. Spontaneous self-assembly of pathogenic huntingtin exon 1 protein into amyloid structures. Essays Biochem. 2014;56:167–80. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
