

Pharmacological targeting of P300/CBP reveals EWS::FLI1-mediated senescence evasion in Ewing sarcoma
- PMID: 39367409
- PMCID: PMC11453018
- DOI: 10.1186/s12943-024-02115-7
Pharmacological targeting of P300/CBP reveals EWS::FLI1-mediated senescence evasion in Ewing sarcoma
Abstract
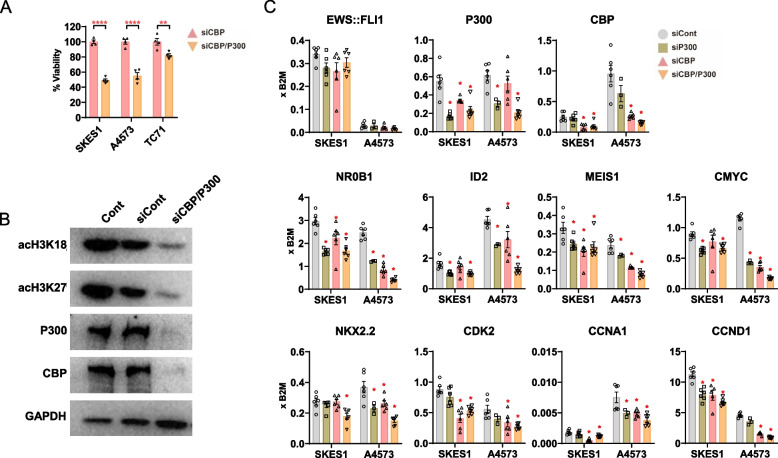
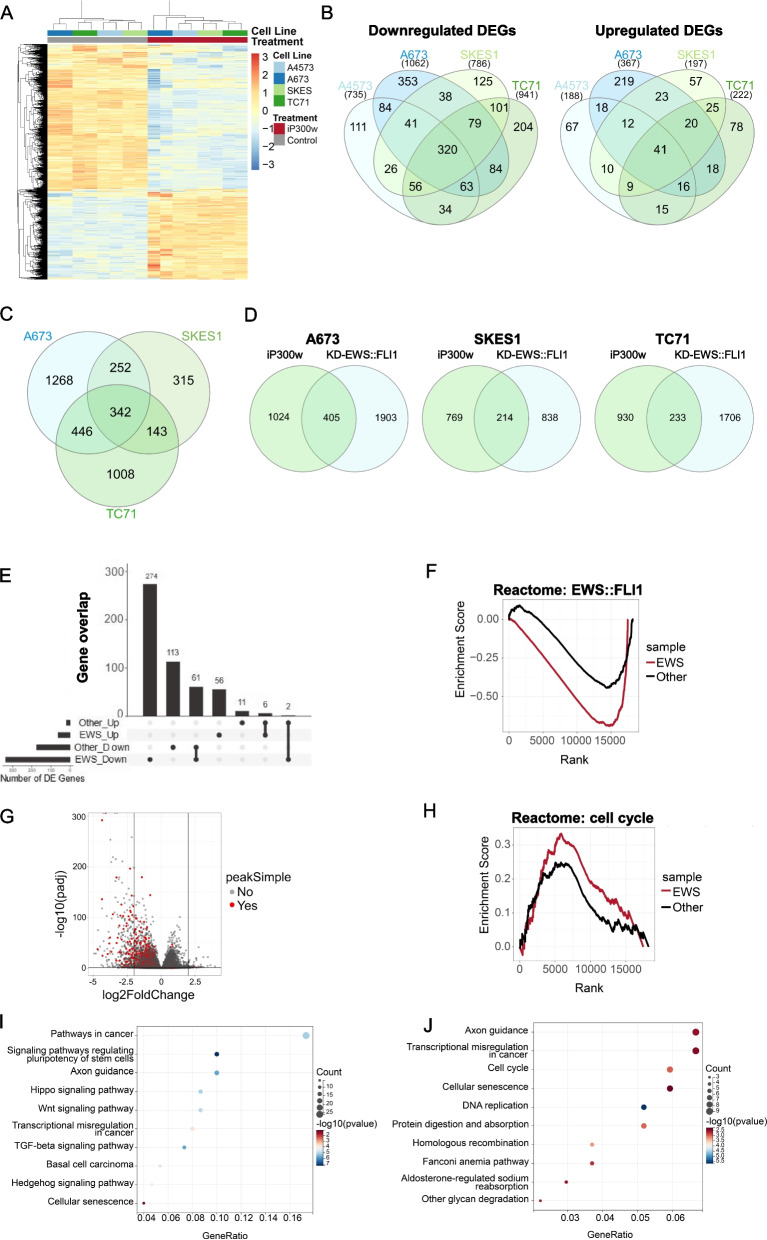
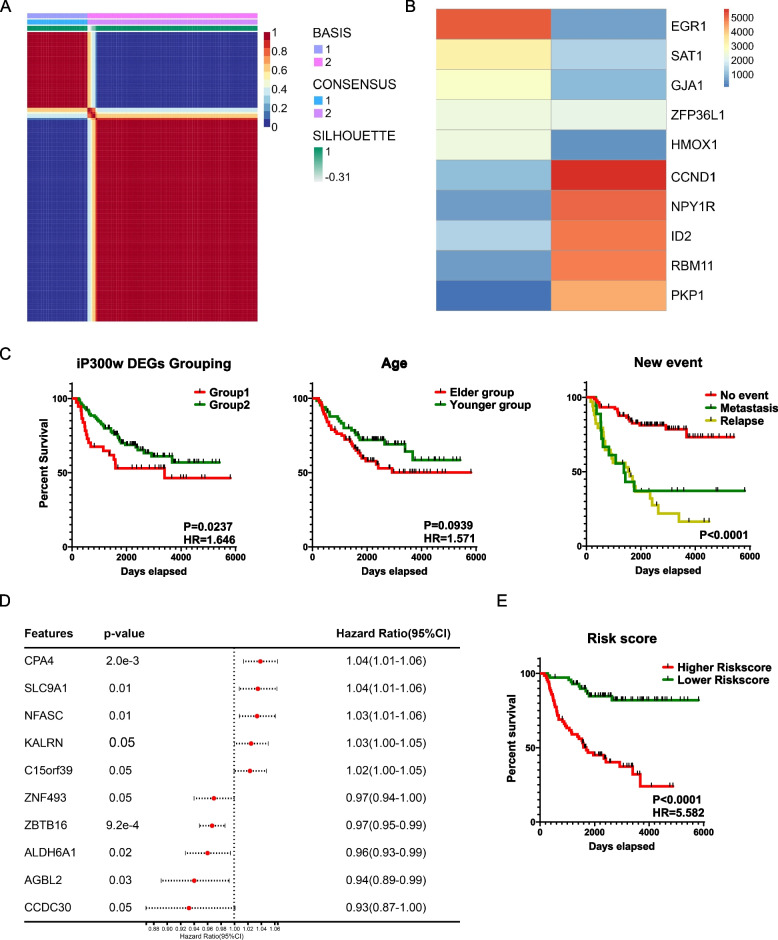
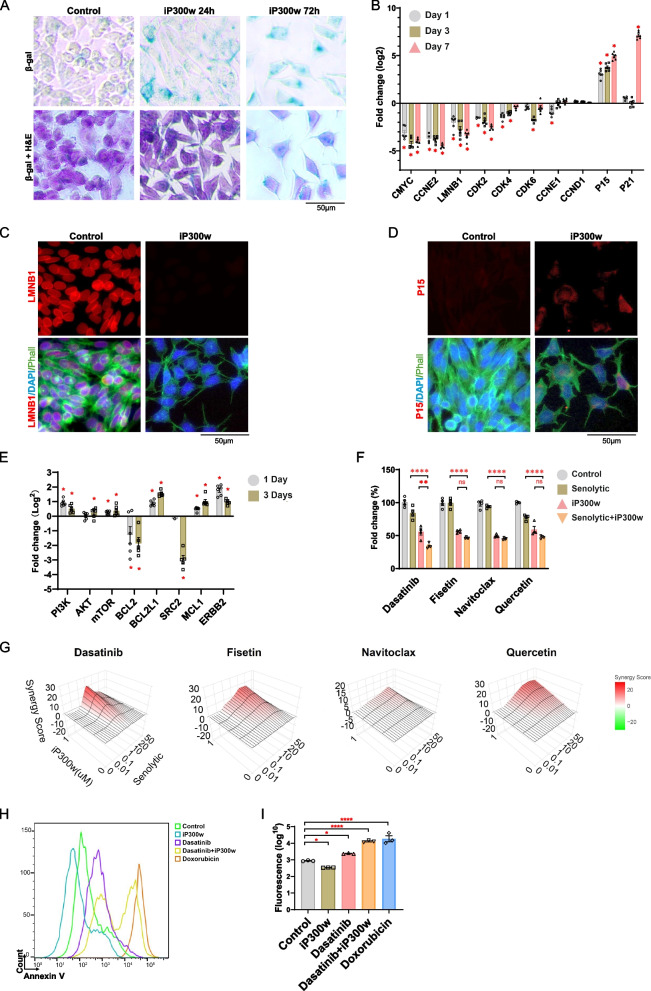
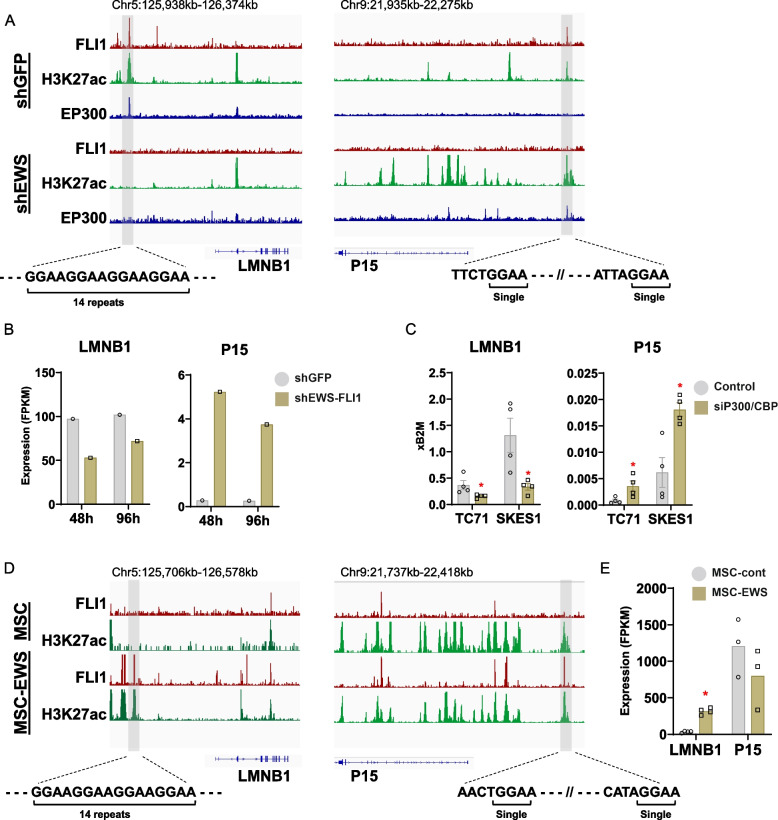
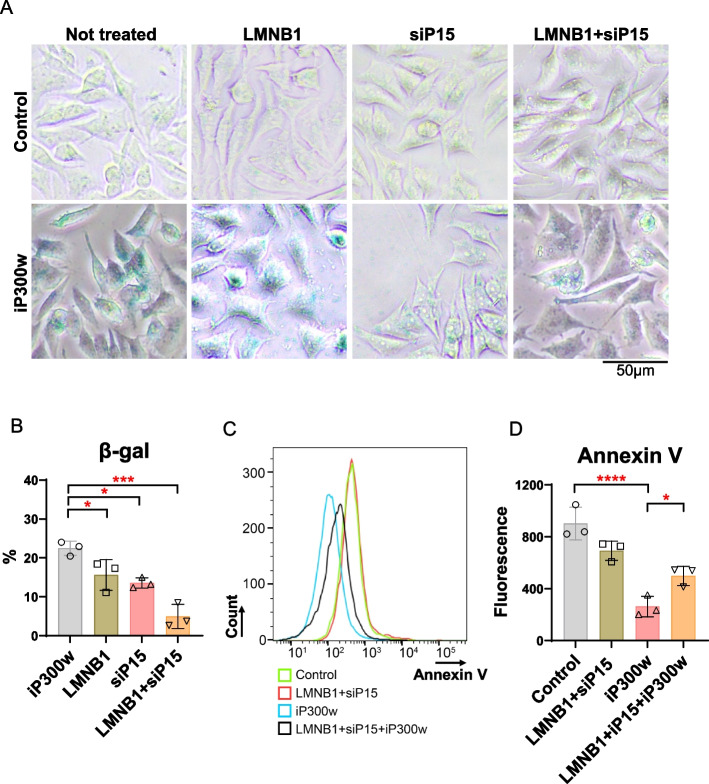
Ewing sarcoma (ES) poses a significant therapeutic challenge due to the difficulty in targeting its main oncodriver, EWS::FLI1. We show that pharmacological targeting of the EWS::FLI1 transcriptional complex via inhibition of P300/CBP drives a global transcriptional outcome similar to direct knockdown of EWS::FLI1, and furthermore yields prognostic risk factors for ES patient outcome. We find that EWS::FLI1 upregulates LMNB1 via repetitive GGAA motif recognition and acetylation codes in ES cells and EWS::FLI1-permissive mesenchymal stem cells, which when reversed by P300 inhibition leads to senescence of ES cells. P300-inhibited senescent ES cells can then be eliminated by senolytics targeting the PI3K signaling pathway. The vulnerability of ES cells to this combination therapy suggests an appealing synergistic strategy for future therapeutic exploration.
Keywords: EWS::FLI1; Ewing sarcoma; Lamin B1; P300/CBP; Pharmacological targeting; Senescence; Senolytics.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Jurgens H, Dirksen U. Ewing sarcoma treatment. Eur J Cancer. 2011;47(Suppl 3):S366–7. - PubMed
-
- Esiashvili N, Goodman M, Marcus RB Jr. Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: surveillance epidemiology and end results data. J Pediatr Hematol Oncol. 2008;30(6):425–30. - PubMed
-
- Subbiah V, et al. Ewing’s sarcoma: standard and experimental treatment options. Curr Treat Options Oncol. 2009;10(1–2):126–40. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
