

Helicobacter pylori PldA modulates TNFR1-mediated p38 signaling pathways to regulate macrophage responses for its survival
- PMID: 39369445
- PMCID: PMC11457642
- DOI: 10.1080/19490976.2024.2409924
Helicobacter pylori PldA modulates TNFR1-mediated p38 signaling pathways to regulate macrophage responses for its survival
Abstract
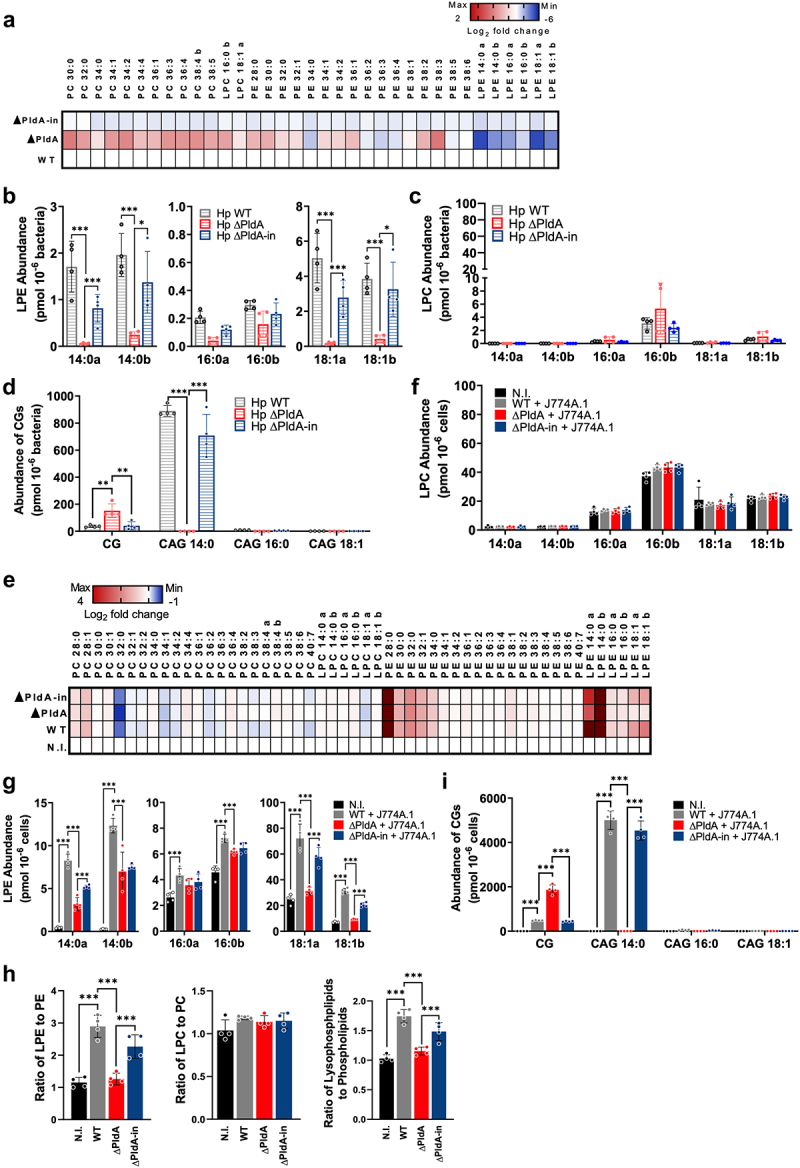
Helicobacter pylori, a dominant member of the gastric microbiota was associated with various gastrointestinal diseases and presents a significant challenge due to increasing antibiotic resistance. This study identifies H. pylori's phospholipase A (PldA) as a critical factor in modulating host macrophage responses, facilitating H. pylori 's evasion of the immune system and persistence. PldA alters membrane lipids through reversible acylation and deacylation, affecting their structure and function. We found that PldA incorporates lysophosphatidylethanolamine into macrophage membranes, disrupting their bilayer structure and impairing TNFR1-mediated p38-MK2 signaling. This disruption results in reduced macrophage autophagy and elevated RIP1-dependent apoptosis, thereby enhancing H. pylori survival, a mechanism also observed in multidrug-resistant strains. Pharmacological inhibition of PldA significantly decreases H. pylori viability and increases macrophage survival. In vivo studies corroborate PldA's essential role in H. pylori persistence and immune cell recruitment. Our findings position PldA as a pivotal element in H. pylori pathogenesis through TNFR1-mediated membrane modulation, offering a promising therapeutic target to counteract bacterial resistance.
Keywords: H. pylori phospholipase A; Host-pathogen interaction; TNFR1 signaling; innate immunity; membrane fluidity.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures









References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous