

This is a preprint.
Allelic effects on KLHL17 expression likely mediated by JunB/D underlie a PDAC GWAS signal at chr1p36.33
- PMID: 39371158
- PMCID: PMC11451706
- DOI: 10.1101/2024.09.16.24313748
Allelic effects on KLHL17 expression likely mediated by JunB/D underlie a PDAC GWAS signal at chr1p36.33
Update in
-
Allelic effects on KLHL17 expression underlie a pancreatic cancer genome-wide association signal at chr1p36.33.Nat Commun. 2025 Apr 30;16(1):4055. doi: 10.1038/s41467-025-59109-2. Nat Commun. 2025. PMID: 40307206 Free PMC article.
Abstract
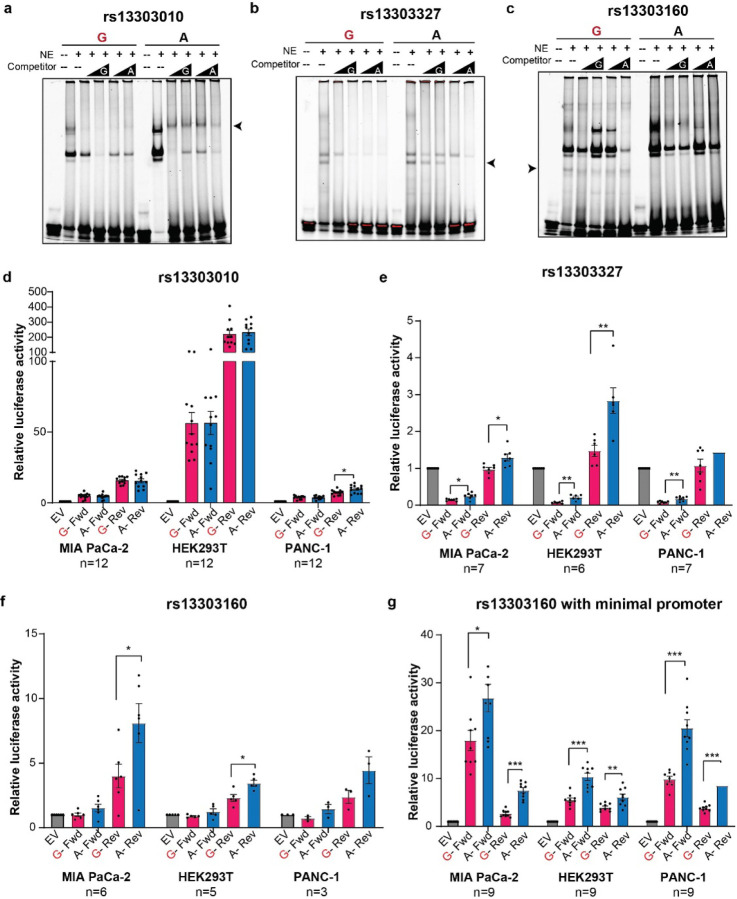
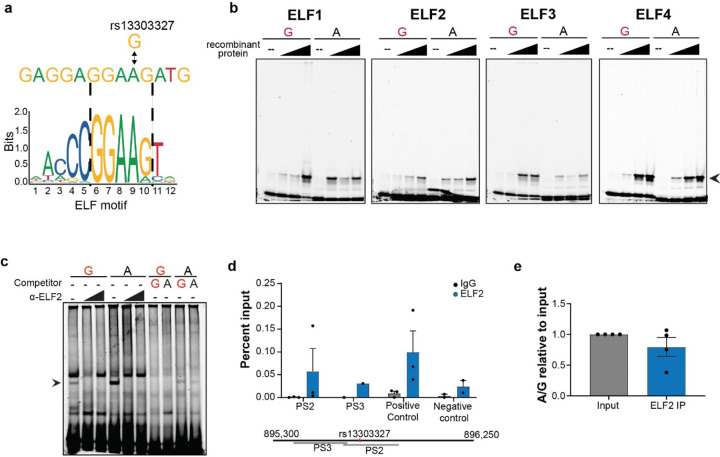
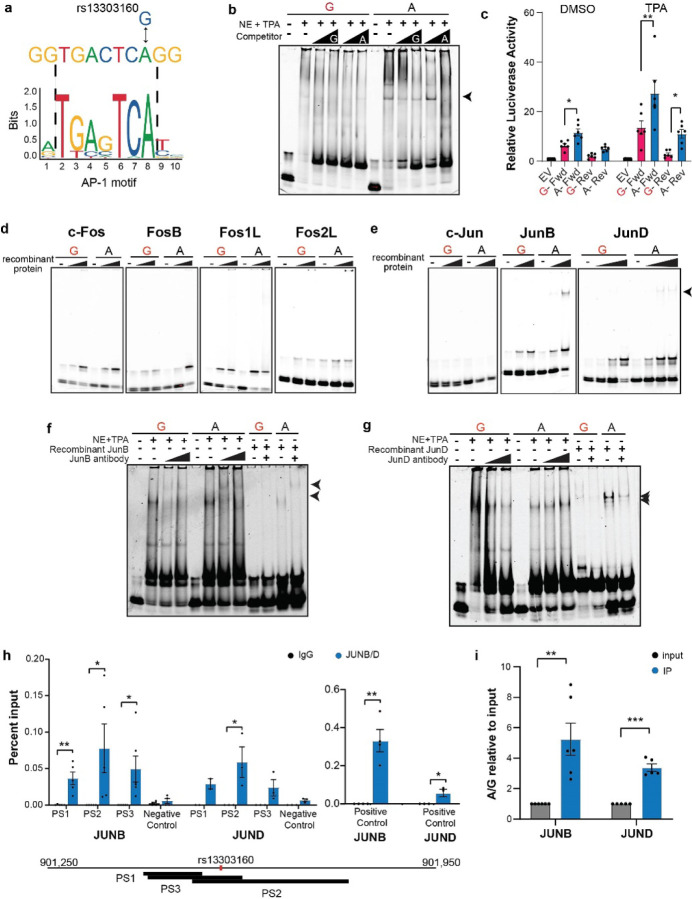
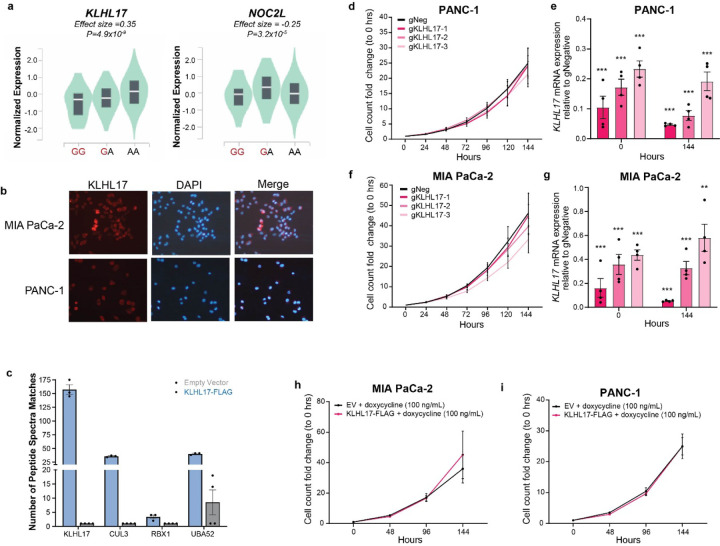
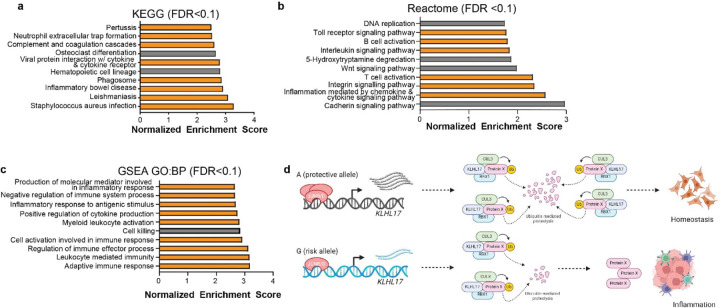
Pancreatic Ductal Adenocarcinoma (PDAC) is the third leading cause of cancer-related deaths in the U.S. Both rare and common germline variants contribute to PDAC risk. Here, we fine-map and functionally characterize a common PDAC risk signal at 1p36.33 (tagged by rs13303010) identified through a genome wide association study (GWAS). One of the fine-mapped SNPs, rs13303160 (r2=0.93 in 1000G EUR samples, OR=1.23, P value=2.74x10-9) demonstrated allele-preferential gene regulatory activity in vitro and allele-preferential binding of JunB and JunD in vitro and in vivo. Expression Quantitative Trait Locus (eQTL) analysis identified KLHL17 as a likely target gene underlying the signal. Proteomic analysis identified KLHL17 as a member of the Cullin-E3 ubiquitin ligase complex in PDAC-derived cells. In silico differential gene expression analysis of the GTExv8 pancreas data suggested an association between lower KLHL17 (risk associated) and pro-inflammatory pathways. We hypothesize that KLHL17 may mitigate inflammation by recruiting pro-inflammatory proteins for ubiquitination and degradation thereby influencing PDAC risk.
Conflict of interest statement
Competing Interests statement The authors declare no competing interests.
Figures

References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources