

This is a preprint.
Exome wide association study for blood lipids in 1,158,017 individuals from diverse populations
- PMID: 39371182
- PMCID: PMC11451673
- DOI: 10.1101/2024.09.17.24313718
Exome wide association study for blood lipids in 1,158,017 individuals from diverse populations
Abstract
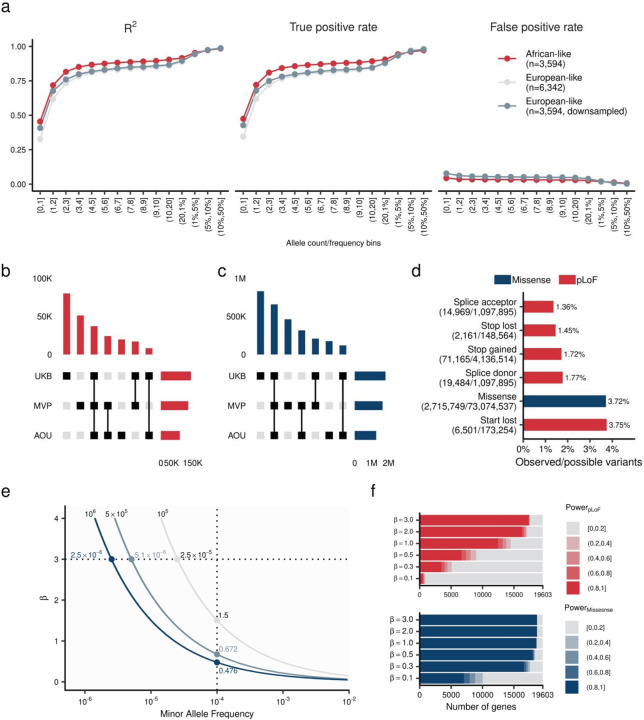
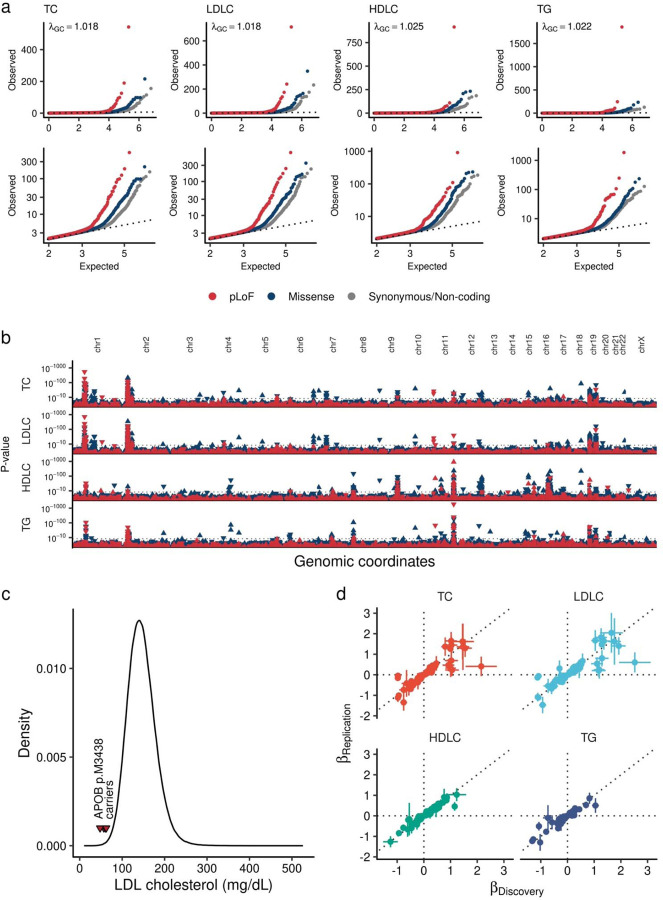
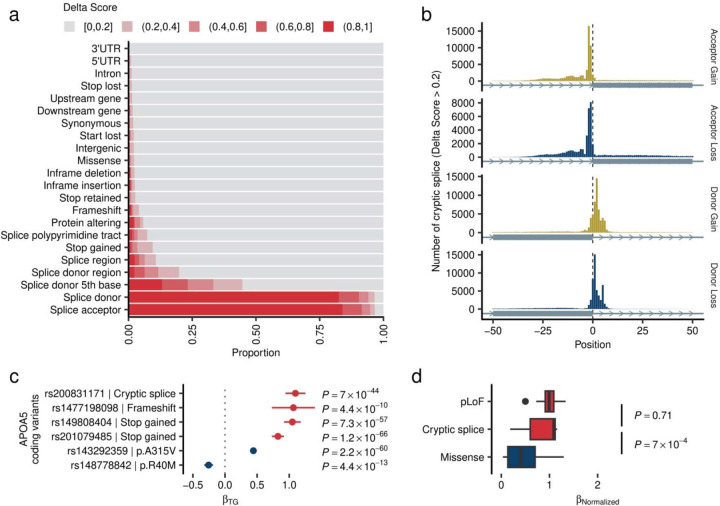
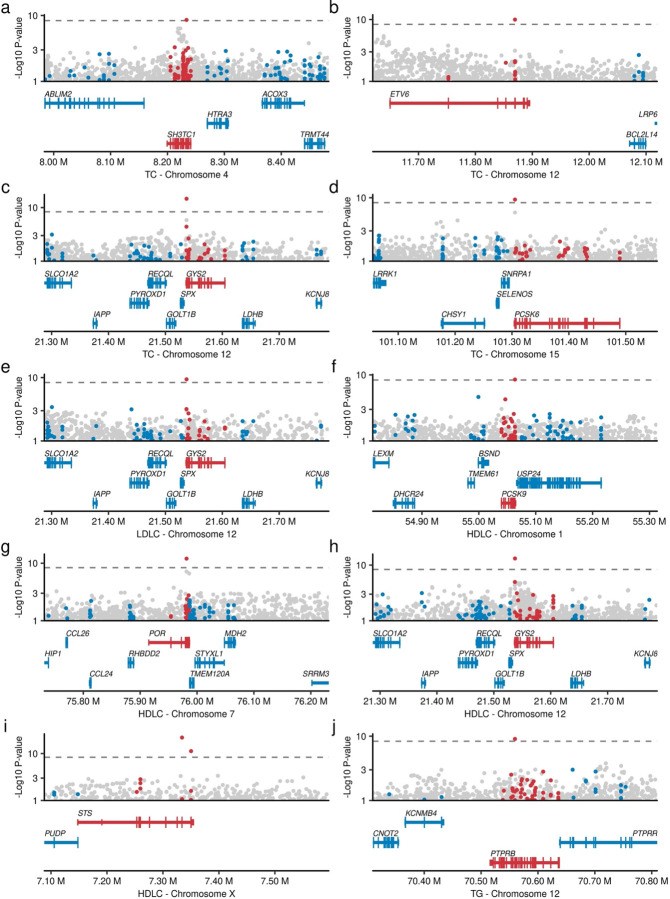
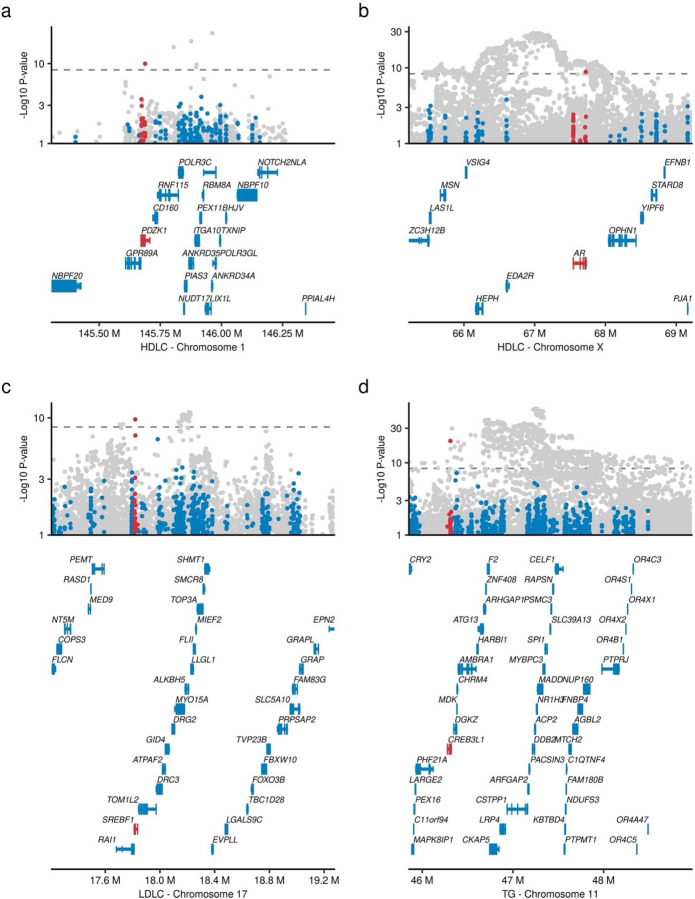
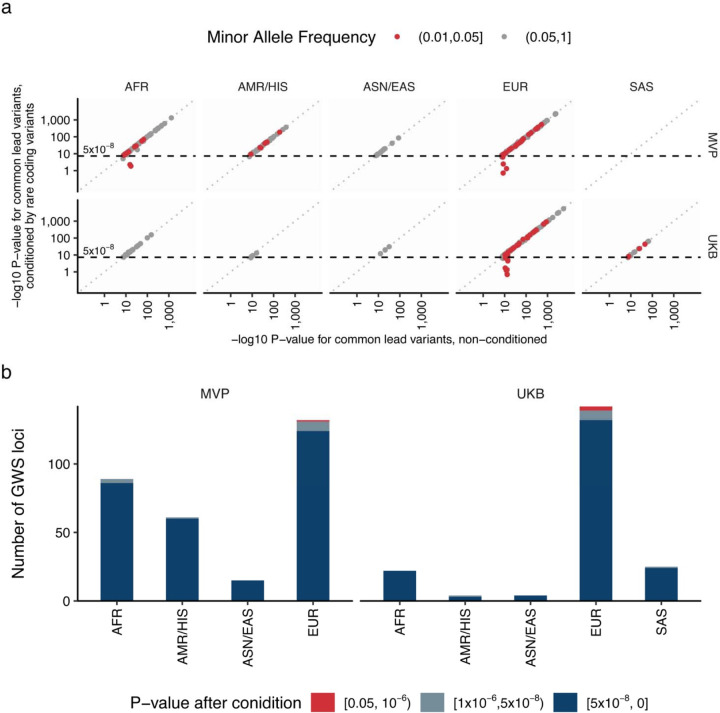
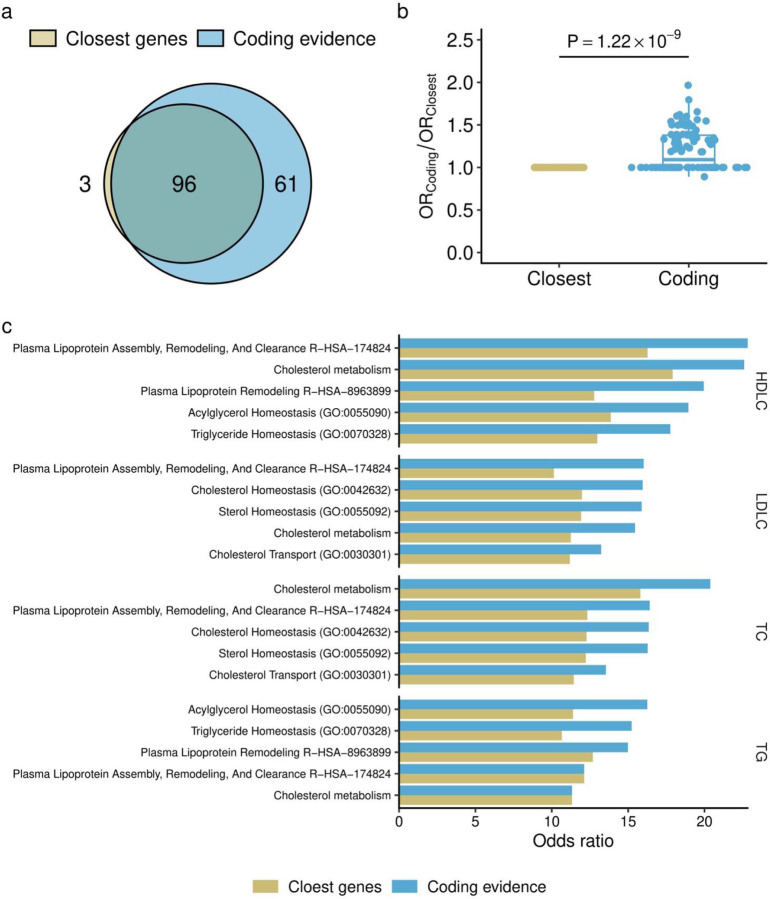
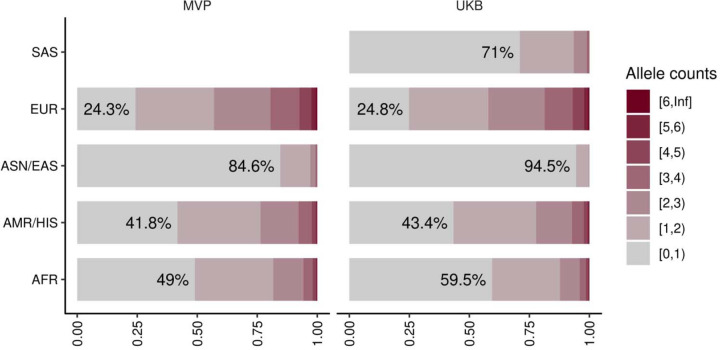
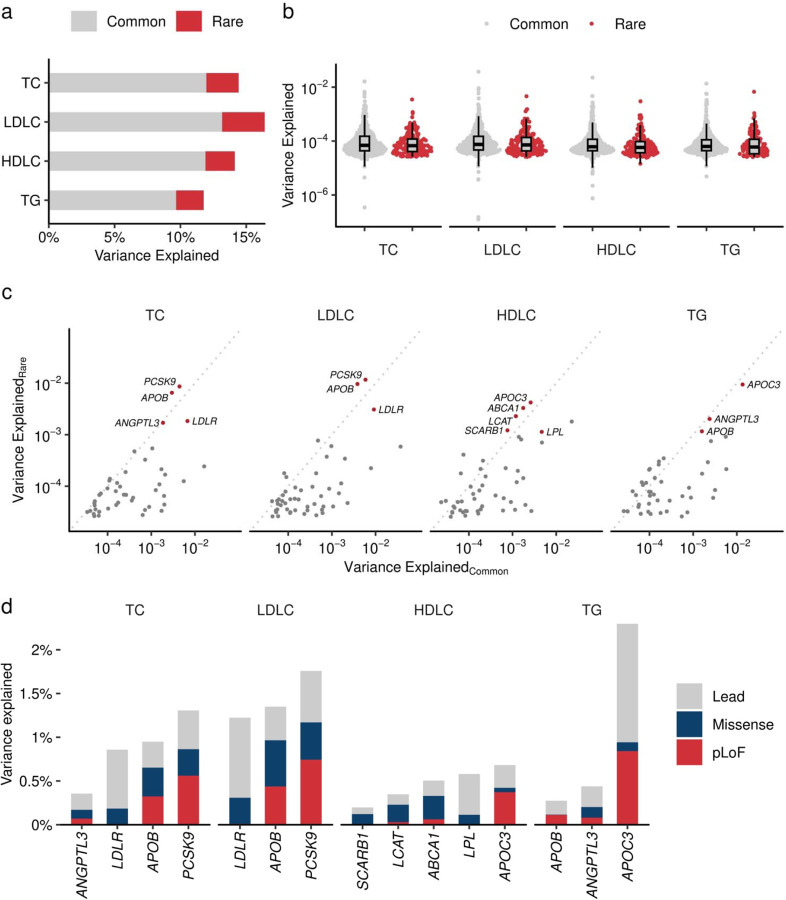
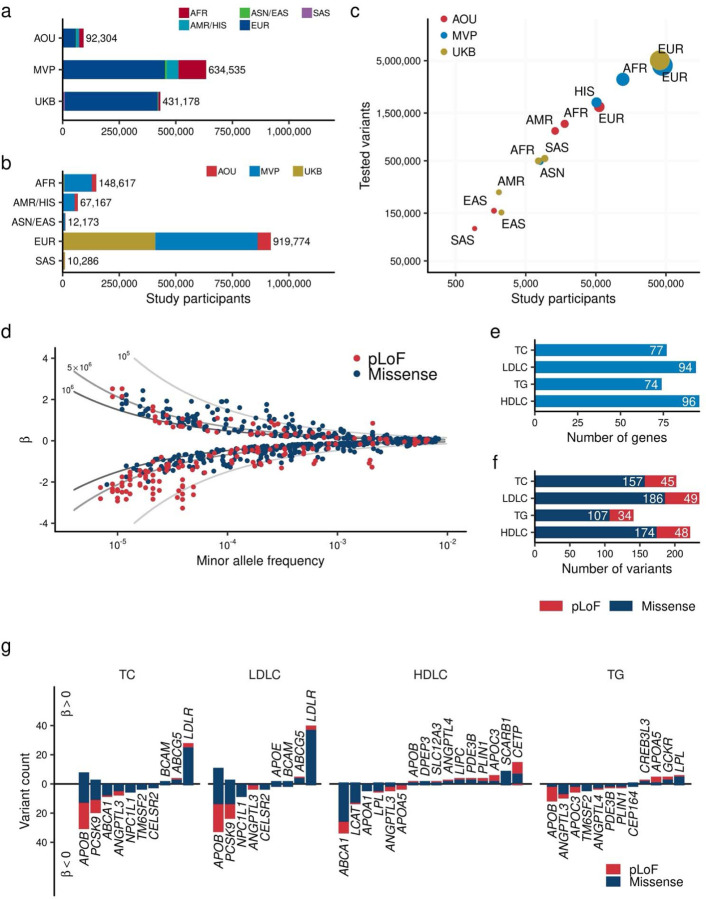
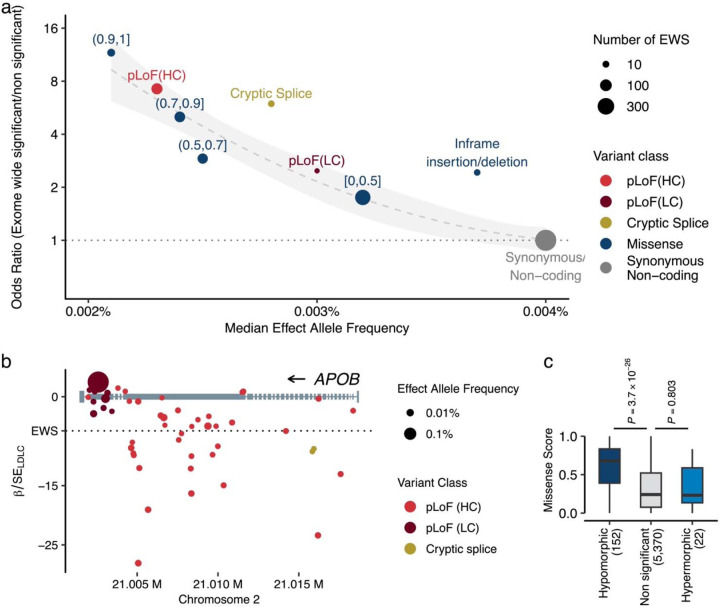
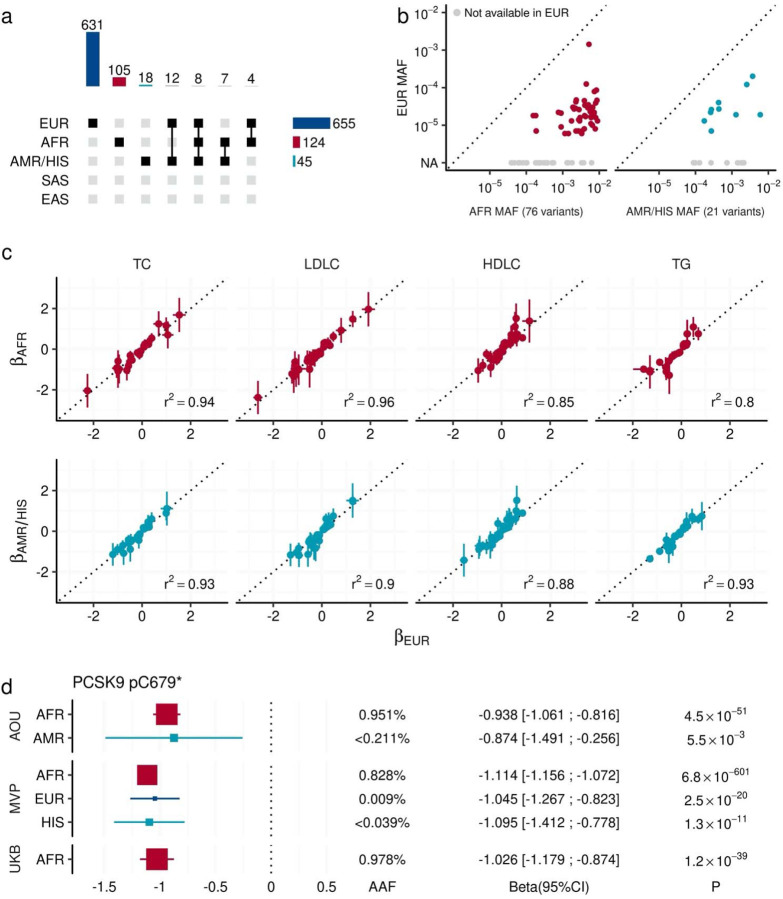
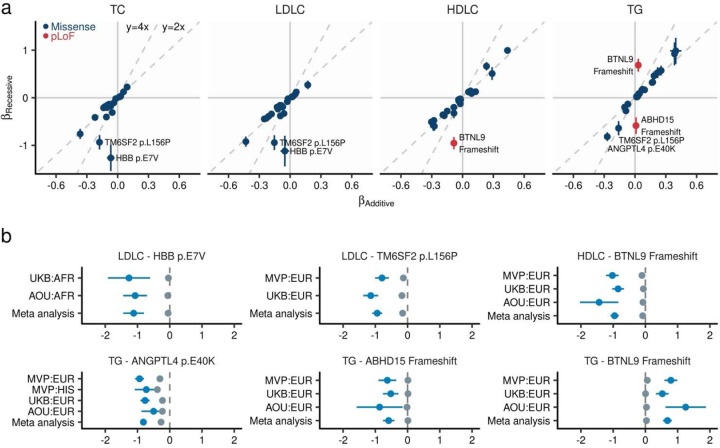
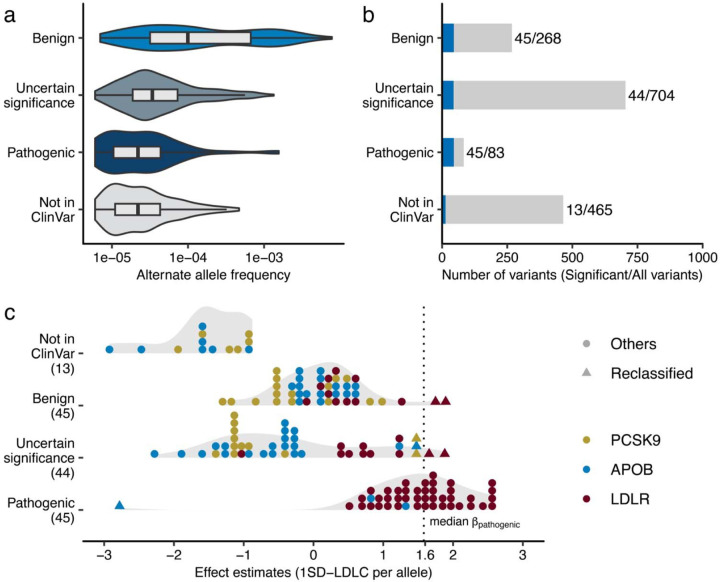
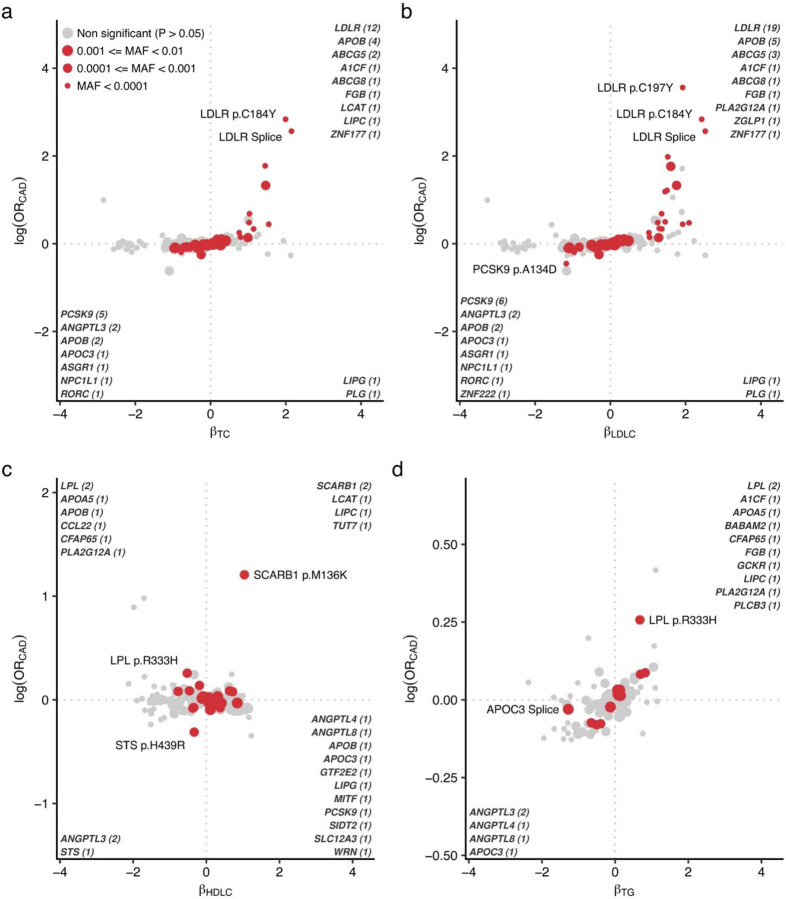
Rare coding alleles play crucial roles in the molecular diagnosis of genetic diseases. However, the systemic identification of these alleles has been challenging due to their scarcity in the general population. Here, we discovered and characterized rare coding alleles contributing to genetic dyslipidemia, a principal risk for coronary artery disease, among over a million individuals combining three large contemporary genetic datasets (the Million Veteran Program, n = 634,535, UK Biobank, n = 431,178, and the All of Us Research Program, n = 92,304) totaling 1,158,017 multi-ancestral individuals. Unlike previous rare variant studies in lipids, this study included 238,243 individuals (20.6%) from non-European-like populations. Testing 2,997,401 rare coding variants from diverse backgrounds, we identified 800 exome-wide significant associations across 209 genes including 176 predicted loss of function and 624 missense variants. Among these exome-wide associations, 130 associations were driven by non-European-like populations. Associated alleles are highly enriched in functional variant classes, showed significant additive and recessive associations, exhibited similar effects across populations, and resolved pathogenicity for variants enriched in African or South-Asian populations. Furthermore, we identified 5 lipid-related genes associated with coronary artery disease (RORC, CFAP65, GTF2E2, PLCB3, and ZNF117). Among them, RORC is a potentially novel therapeutic target through the down regulation of LDLC by its silencing. This study provides resources and insights for understanding causal mechanisms, quantifying the expressivity of rare coding alleles, and identifying novel drug targets across diverse populations.
Conflict of interest statement
Competing interest declaration D.K. is a scientific advisor and reports consulting fees from Bitterroot Bio, Inc unrelated to the present work. P.N. reports research grants from Allelica, Amgen, Apple, Boston Scientific, Genentech / Roche, and Novartis, personal fees from Allelica, Apple, AstraZeneca, Blackstone Life Sciences, Creative Education Concepts, CRISPR Therapeutics, Eli Lilly & Co, Foresite Labs, Genentech / Roche, GV, HeartFlow, Magnet Biomedicine, Merck, and Novartis, scientific advisory board membership of Esperion Therapeutics, Preciseli, and TenSixteen Bio, scientific co-founder of TenSixteen Bio, equity in MyOme, Preciseli, and TenSixteen Bio, and spousal employment at Vertex Pharmaceuticals, all unrelated to the present work.
Figures
References
-
- Gidding S.S., et al. The Agenda for Familial Hypercholesterolemia: A Scientific Statement From the American Heart Association. Circulation 132, 2167–2192 (2015). - PubMed
-
- Pijlman A.H., et al. Evaluation of cholesterol lowering treatment of patients with familial hypercholesterolemia: a large cross-sectional study in The Netherlands. Atherosclerosis 209, 189–194 (2010). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources