

This is a preprint.
REAL-TIME VISUALIZATION OF SPLICEOSOME ASSEMBLY REVEALS BASIC PRINCIPLES OF SPLICE SITE SELECTION
- PMID: 39372787
- PMCID: PMC11451613
- DOI: 10.1101/2024.07.12.603320
REAL-TIME VISUALIZATION OF SPLICEOSOME ASSEMBLY REVEALS BASIC PRINCIPLES OF SPLICE SITE SELECTION
Abstract
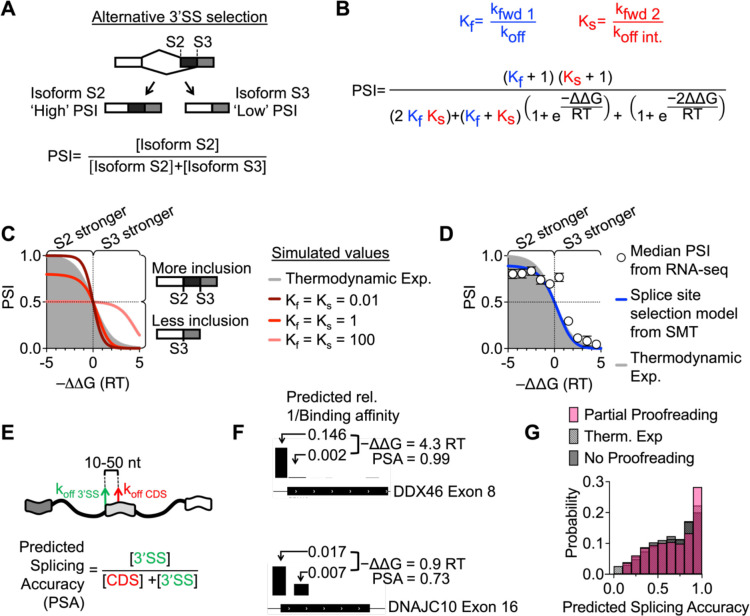
The spliceosome is a megadalton protein-RNA complex which removes introns from pre-mRNA, yet the dynamic early assembly steps have not been structurally resolved. Specifically, how the spliceosome selects the correct 3' splice site (3'SS) amongst highly similar non-functional sites is not known. Here, we develop a kinetic model of splice site selection based on single-molecule U2AF heterodimer imaging in vitro and in vivo. The model successfully predicts alternative splicing patterns and indicates that 3'SS selection occurs while U2AF is in complex with the spliceosome, not during initial binding. This finding indicates the spliceosome operates in a 'partial' kinetic proofreading regime, catalyzed in part by the helicase DDX42, which increases selectivity to the underlying U2AF binding site while still allowing for efficient forward progression.
Figures



References
-
- Jurica M. S., Moore M. J., Pre-mRNA splicing: awash in a sea of proteins. Mol Cell 12, 5–14 (2003). - PubMed
Publication types
LinkOut - more resources
Full Text Sources