

This is a preprint.
Complex genetic variation in nearly complete human genomes
- PMID: 39372794
- PMCID: PMC11451754
- DOI: 10.1101/2024.09.24.614721
Complex genetic variation in nearly complete human genomes
Update in
-
Complex genetic variation in nearly complete human genomes.Nature. 2025 Aug;644(8076):430-441. doi: 10.1038/s41586-025-09140-6. Epub 2025 Jul 23. Nature. 2025. PMID: 40702183 Free PMC article.
Abstract
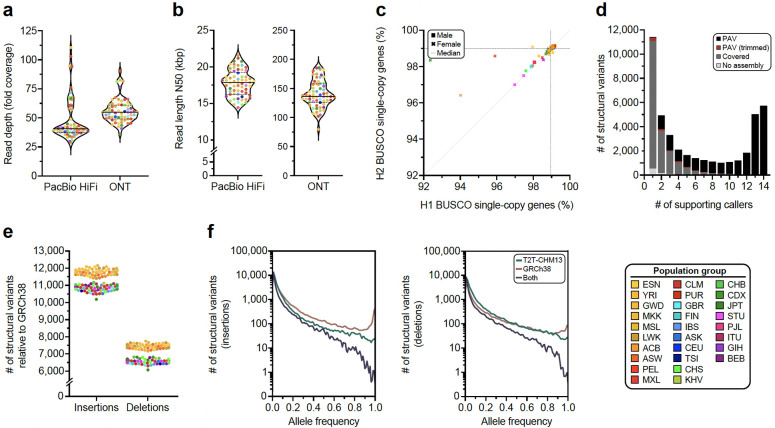
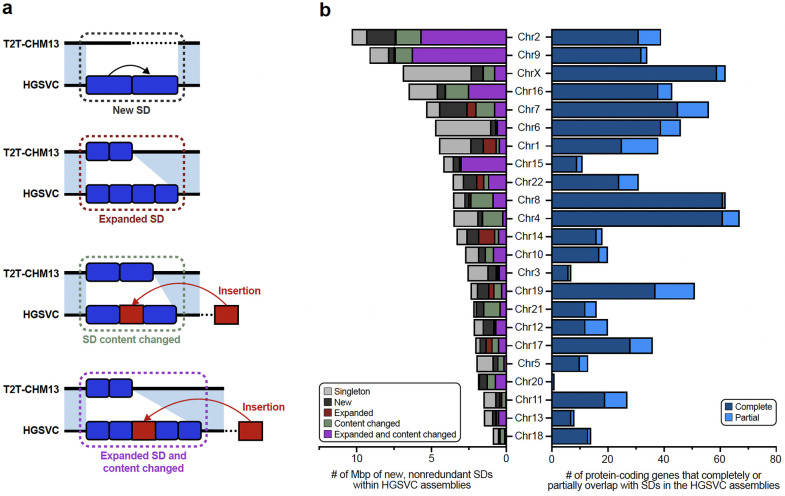
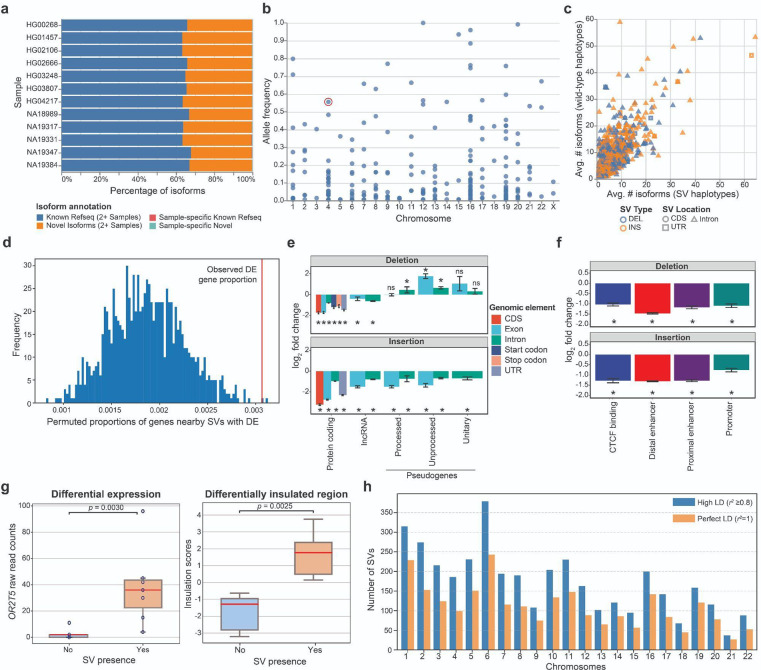
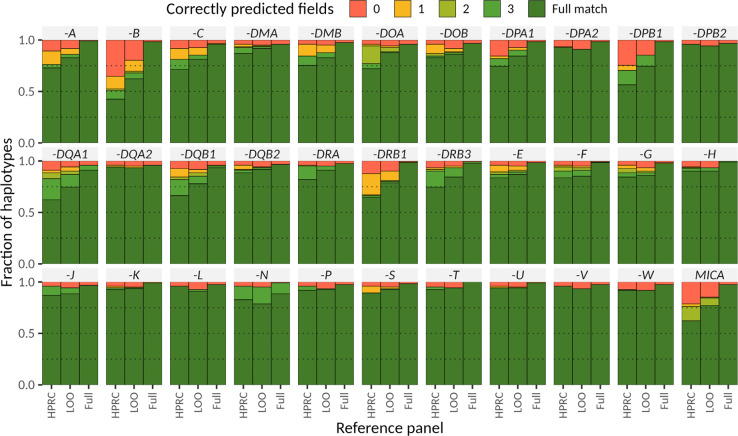
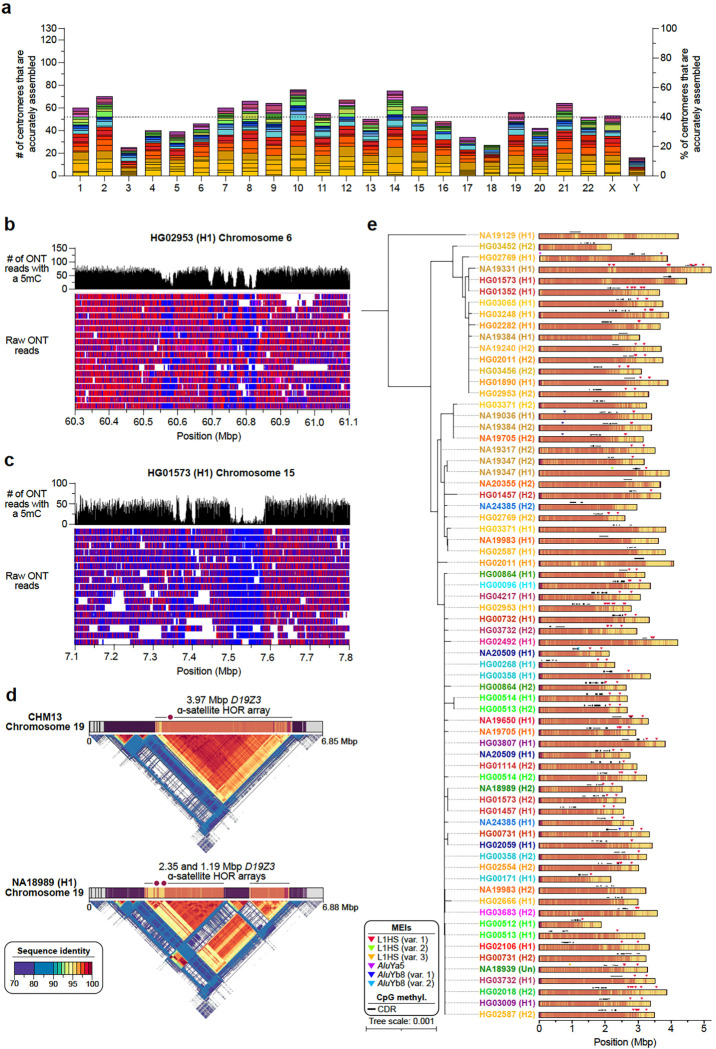
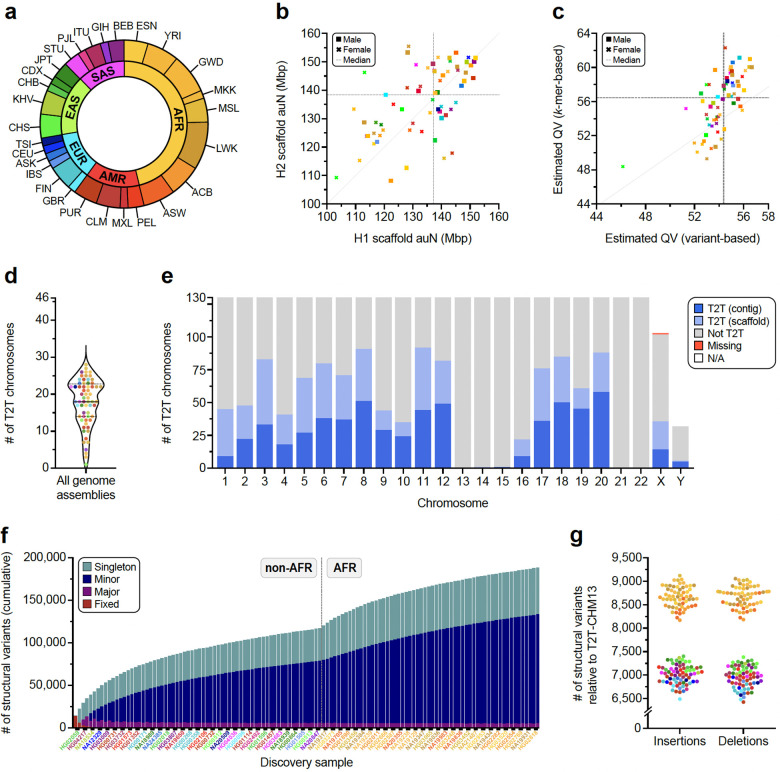
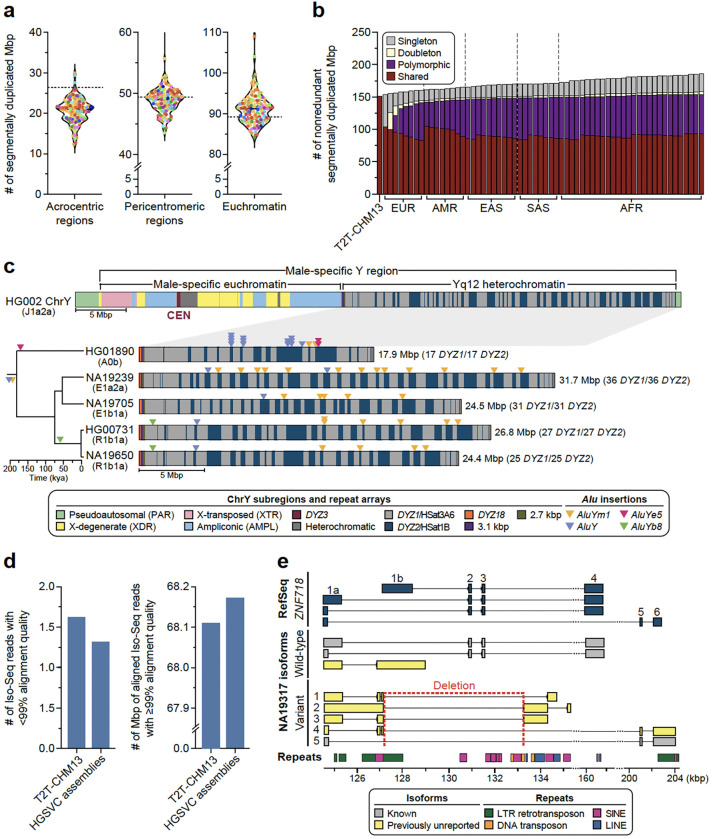
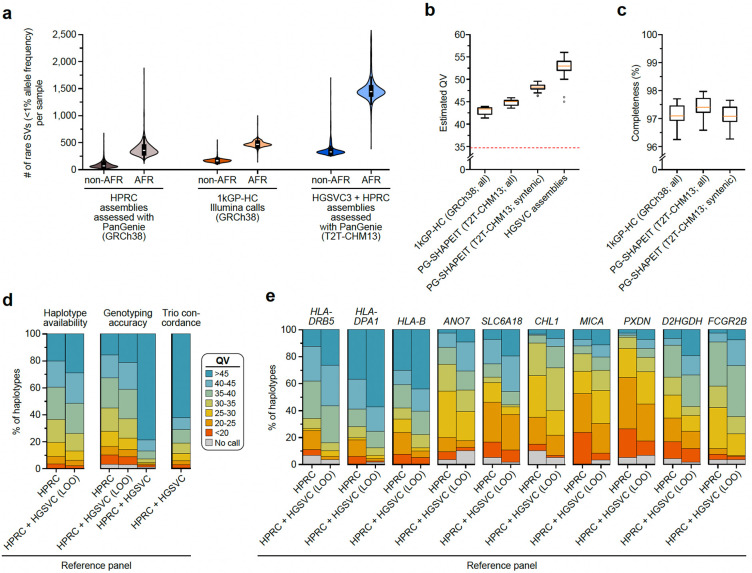
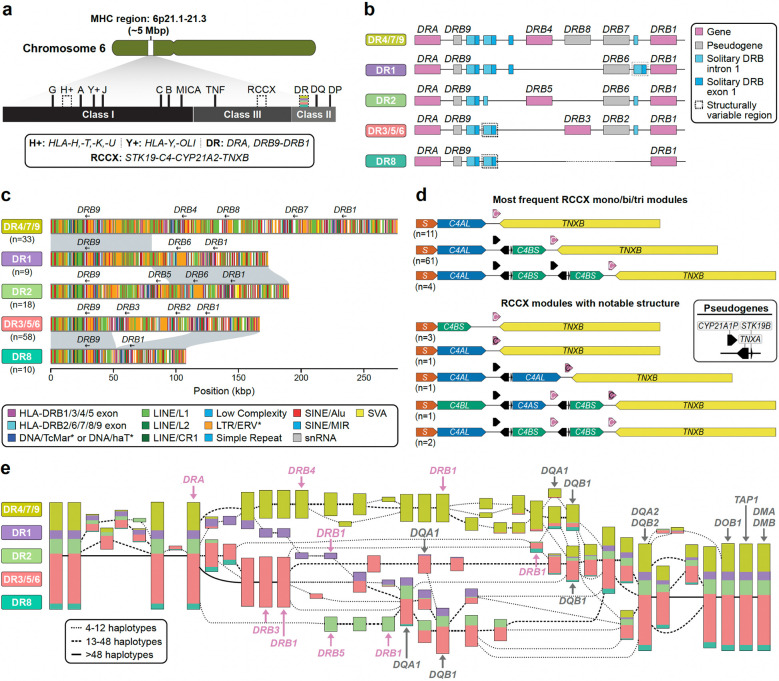
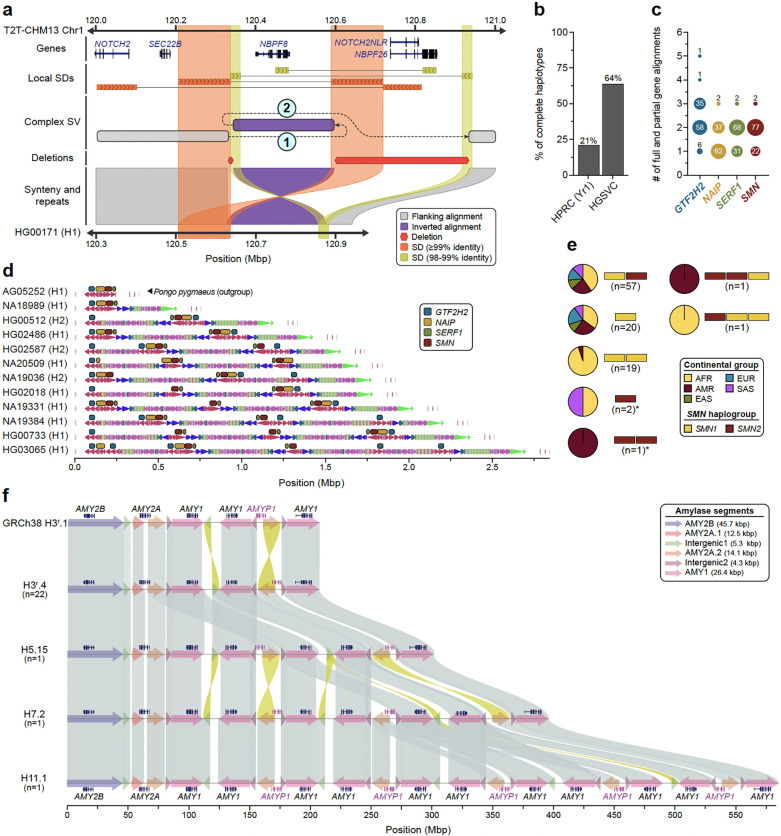
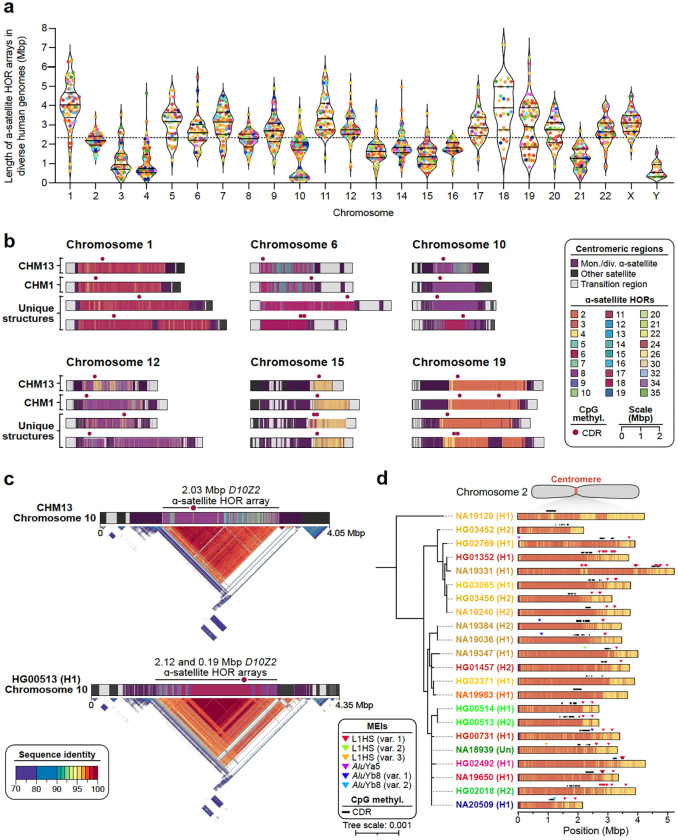
Diverse sets of complete human genomes are required to construct a pangenome reference and to understand the extent of complex structural variation. Here, we sequence 65 diverse human genomes and build 130 haplotype-resolved assemblies (130 Mbp median continuity), closing 92% of all previous assembly gaps1,2 and reaching telomere-to-telomere (T2T) status for 39% of the chromosomes. We highlight complete sequence continuity of complex loci, including the major histocompatibility complex (MHC), SMN1/SMN2, NBPF8, and AMY1/AMY2, and fully resolve 1,852 complex structural variants (SVs). In addition, we completely assemble and validate 1,246 human centromeres. We find up to 30-fold variation in α-satellite high-order repeat (HOR) array length and characterize the pattern of mobile element insertions into α-satellite HOR arrays. While most centromeres predict a single site of kinetochore attachment, epigenetic analysis suggests the presence of two hypomethylated regions for 7% of centromeres. Combining our data with the draft pangenome reference1 significantly enhances genotyping accuracy from short-read data, enabling whole-genome inference3 to a median quality value (QV) of 45. Using this approach, 26,115 SVs per sample are detected, substantially increasing the number of SVs now amenable to downstream disease association studies.
Conflict of interest statement
Competing Interests E.E.E. is a scientific advisory board member of Variant Bio, Inc. C. Lee is a scientific advisory board member of Nabsys and Genome Insight. S.K. has received travel funds to speak at events hosted by Oxford Nanopore Technologies. The following authors have previously disclosed a patent application (No. EP19169090) relevant to Strand-seq: J.O.K., T.M., and D.P. The other authors declare no competing interests.
Figures
References
-
- Liao W-W, Asri M, Ebler J, Doerr D, Haukness M, Hickey G, Lu S, Lucas JK, Monlong J, Abel HJ, Buonaiuto S, Chang XH, Cheng H, Chu J, Colonna V, Eizenga JM, Feng X, Fischer C, Fulton RS, Garg S, Groza C, Guarracino A, Harvey WT, Heumos S, Howe K, Jain M, Lu T-Y, Markello C, Martin FJ, Mitchell MW, Munson KM, Mwaniki MN, Novak AM, Olsen HE, Pesout T, Porubsky D, Prins P, Sibbesen JA, Sirén J, Tomlinson C, Villani F, Vollger MR, Antonacci-Fulton LL, Baid G, Baker CA, Belyaeva A, Billis K, Carroll A, Chang P-C, Cody S, Cook DE, Cook-Deegan RM, Cornejo OE, Diekhans M, Ebert P, Fairley S, Fedrigo O, Felsenfeld AL, Formenti G, Frankish A, Gao Y, Garrison NA, Giron CG, Green RE, Haggerty L, Hoekzema K, Hourlier T, Ji HP, Kenny EE, Koenig BA, Kolesnikov A, Korbel JO, Kordosky J, Koren S, Lee H, Lewis AP, Magalhães H, Marco-Sola S, Marijon P, McCartney A, McDaniel J, Mountcastle J, Nattestad M, Nurk S, Olson ND, Popejoy AB, Puiu D, Rautiainen M, Regier AA, Rhie A, Sacco S, Sanders AD, Schneider VA, Schultz BI, Shafin K, Smith MW, Sofia HJ, Abou Tayoun AN, et al. A draft human pangenome reference. Nature. 2023;617:312–324. - PMC - PubMed
-
- Porubsky D, Vollger MR, Harvey WT, Rozanski AN, Ebert P, Hickey G, Hasenfeld P, Sanders AD, Stober C, Human Pangenome Reference Consortium, Korbel JO, Paten B, Marschall T, Eichler EE. Gaps and complex structurally variant loci in phased genome assemblies. Genome Res. 2023;33:496–510. - PMC - PubMed
-
- Jarvis ED, Formenti G, Rhie A, Guarracino A, Yang C, Wood J, Tracey A, Thibaud-Nissen F, Vollger MR, Porubsky D, Cheng H, Asri M, Logsdon GA, Carnevali P, Chaisson MJP, Chin C-S, Cody S, Collins J, Ebert P, Escalona M, Fedrigo O, Fulton RS, Fulton LL, Garg S, Gerton JL, Ghurye J, Granat A, Green RE, Harvey W, Hasenfeld P, Hastie A, Haukness M, Jaeger EB, Jain M, Kirsche M, Kolmogorov M, Korbel JO, Koren S, Korlach J, Lee J, Li D, Lindsay T, Lucas J, Luo F, Marschall T, Mitchell MW, McDaniel J, Nie F, Olsen HE, Olson ND, Pesout T, Potapova T, Puiu D, Regier A, Ruan J, Salzberg SL, Sanders AD, Schatz MC, Schmitt A, Schneider VA, Selvaraj S, Shafin K, Shumate A, Stitziel NO, Stober C, Torrance J, Wagner J, Wang J, Wenger A, Xiao C, Zimin AV, Zhang G, Wang T, Li H, Garrison E, Haussler D, Hall I, Zook JM, Eichler EE, Phillippy AM, Paten B, Howe K, Miga KH, Human Pangenome Reference Consortium. Semi-automated assembly of high-quality diploid human reference genomes. Nature. 2022;611:519–531. - PMC - PubMed
Publication types
Grants and funding
- R21 CA259309/CA/NCI NIH HHS/United States
- R35 GM138212/GM/NIGMS NIH HHS/United States
- R35 GM133600/GM/NIGMS NIH HHS/United States
- R01 HG011649/HG/NHGRI NIH HHS/United States
- P30 CA034196/CA/NCI NIH HHS/United States
- R01 CA261934/CA/NCI NIH HHS/United States
- R00 HG012798/HG/NHGRI NIH HHS/United States
- K99 GM147352/GM/NIGMS NIH HHS/United States
- K99 HG012798/HG/NHGRI NIH HHS/United States
- U24 HG007497/HG/NHGRI NIH HHS/United States
- P20 GM139769/GM/NIGMS NIH HHS/United States
- R00 GM147352/GM/NIGMS NIH HHS/United States
- U01 AI090905/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous