

Scalable, compressed phenotypic screening using pooled perturbations
- PMID: 39375446
- PMCID: PMC12339377
- DOI: 10.1038/s41587-024-02403-z
Scalable, compressed phenotypic screening using pooled perturbations
Abstract
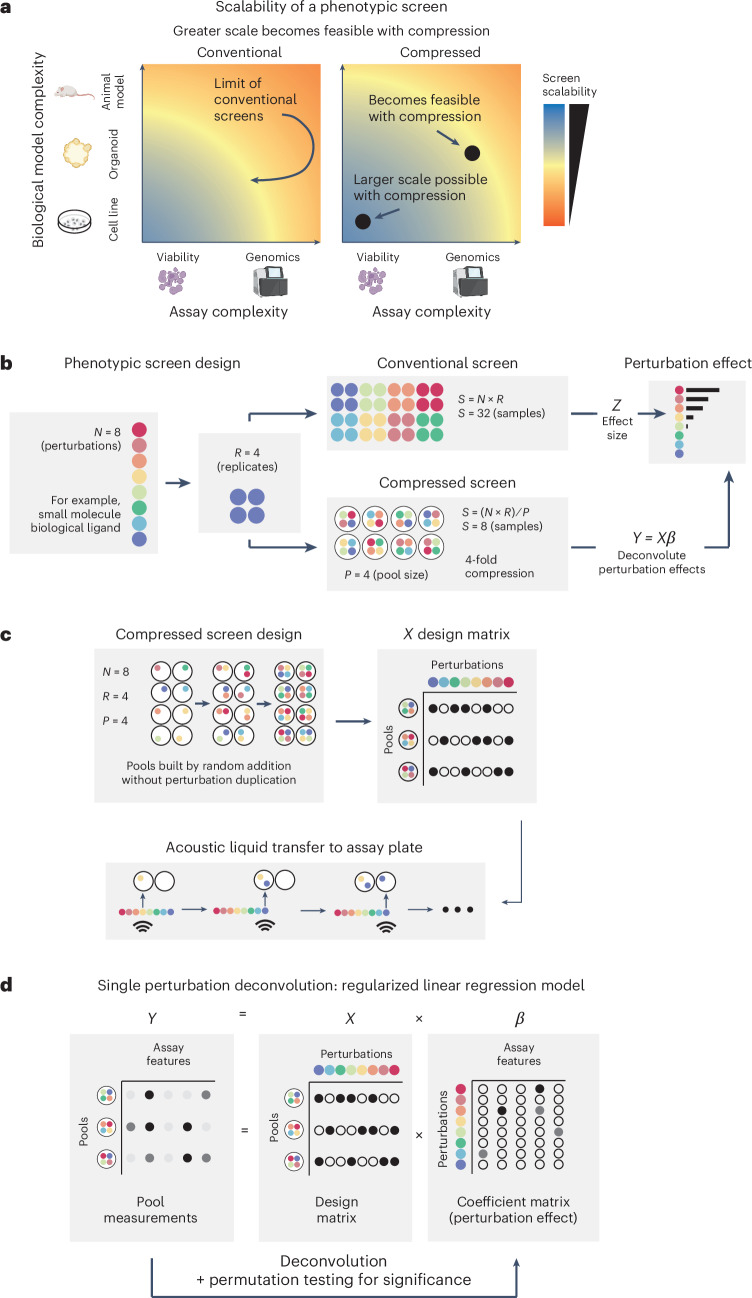
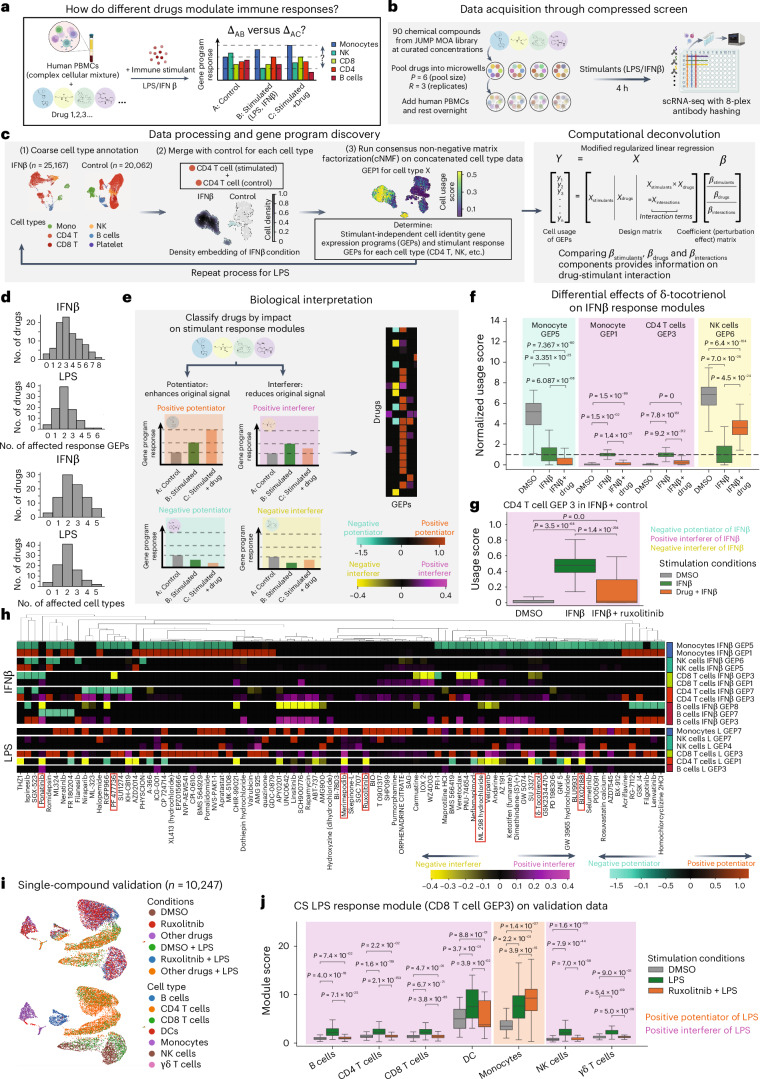
High-throughput phenotypic screens using biochemical perturbations and high-content readouts are constrained by limitations of scale. To address this, we establish a method of pooling exogenous perturbations followed by computational deconvolution to reduce required sample size, labor and cost. We demonstrate the increased efficiency of compressed experimental designs compared to conventional approaches through benchmarking with a bioactive small-molecule library and a high-content imaging readout. We then apply compressed screening in two biological discovery campaigns. In the first, we use early-passage pancreatic cancer organoids to map transcriptional responses to a library of recombinant tumor microenvironment protein ligands, uncovering reproducible phenotypic shifts induced by specific ligands distinct from canonical reference signatures and correlated with clinical outcome. In the second, we identify the pleotropic modulatory effects of a chemical compound library with known mechanisms of action on primary human peripheral blood mononuclear cell immune responses. In sum, our approach empowers phenotypic screens with information-rich readouts to advance drug discovery efforts and basic biological inquiry.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: A.K.S. reports compensation for consulting and/or scientific advisory board (SAB) membership from Honeycomb Biotechnologies, Cellarity, Ochre Bio, Bio-Rad Laboratories, Relation Therapeutics, IntrECate biotherapeutics, Fog Pharma, Passkey Therapeutics and Dahlia Biosciences unrelated to this work. B.E.M. reports compensation for consulting from Empress Therapeutics unrelated to this work. S.R. holds equity in Amgen and receives research funding from Microsoft. P.S.W. receives research funding from Microsoft and reports compensation for consulting from The Engine Venture unrelated to this work. P.C.B. is a consultant to or holds equity in 10X Genomics, General Automation Lab Technologies/Isolation Bio, Celsius Therapeutics, Next Gen Diagnostics, Cache DNA, Concerto Biosciences, Stately, Ramona Optics and Bifrost. W.C.H. is a consultant for Thermo Fisher, Solasta Ventures, KSQ Therapeutics, Jubilant Therapeutics, RAPPTA Therapeutics, Function Oncology, Riva Therapeutics, Serinus Biosciences, Crane Biosciences, Frontier Medicines, Kestrel Therapeutics and Calyx. W.H. reports SAB membership from Kestrel Therapeutics. L.C. and A.P.A. are both employees of Microsoft Research and hold equities in Microsoft. The other authors declare no competing interests.
Figures












References
-
- Joyce, A. R. & Palsson, B. Ø. The model organism as a system: integrating ‘omics’ data sets. Nat. Rev. Mol. Cell Biol.7, 198–210 (2006). - PubMed
-
- Eder, J., Sedrani, R. & Wiesmann, C. The discovery of first-in-class drugs: origins and evolution. Nat. Rev. Drug Discov.13, 577–587 (2014). - PubMed
-
- Swinney, D. C. & Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov.10, 507–519 (2011). - PubMed
-
- Moffat, J. G., Vincent, F., Lee, J. A., Eder, J. & Prunotto, M. Opportunities and challenges in phenotypic drug discovery: an industry perspective. Nat. Rev. Drug Discov.16, 531–543 (2017). - PubMed
-
- Swinney, D. C. Phenotypic vs. target-based drug discovery for first-in-class medicines. Clin. Pharmacol. Ther.93, 299–301 (2013). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
