

Inhibition and degradation of NRAS with a pan-NRAS monobody
- PMID: 39379700
- PMCID: PMC11584388
- DOI: 10.1038/s41388-024-03186-y
Inhibition and degradation of NRAS with a pan-NRAS monobody
Abstract
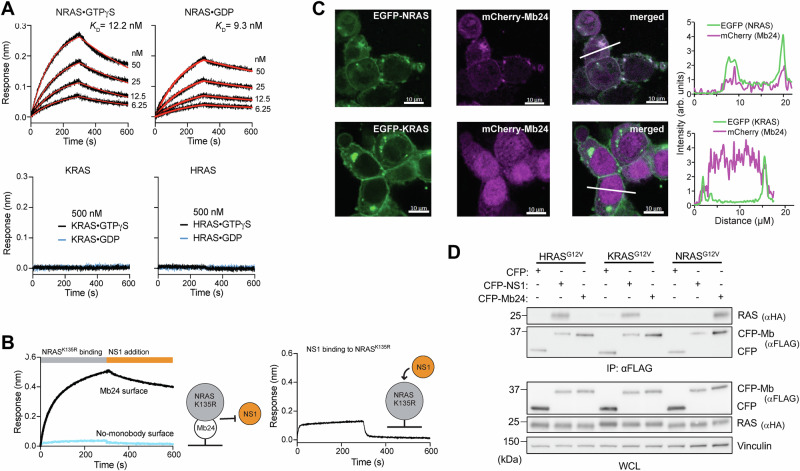
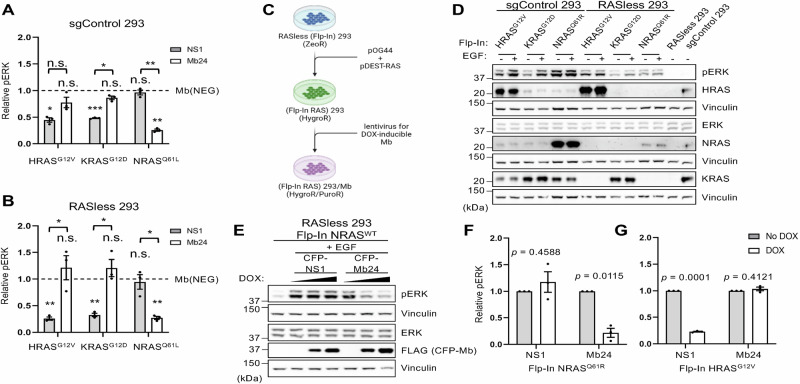
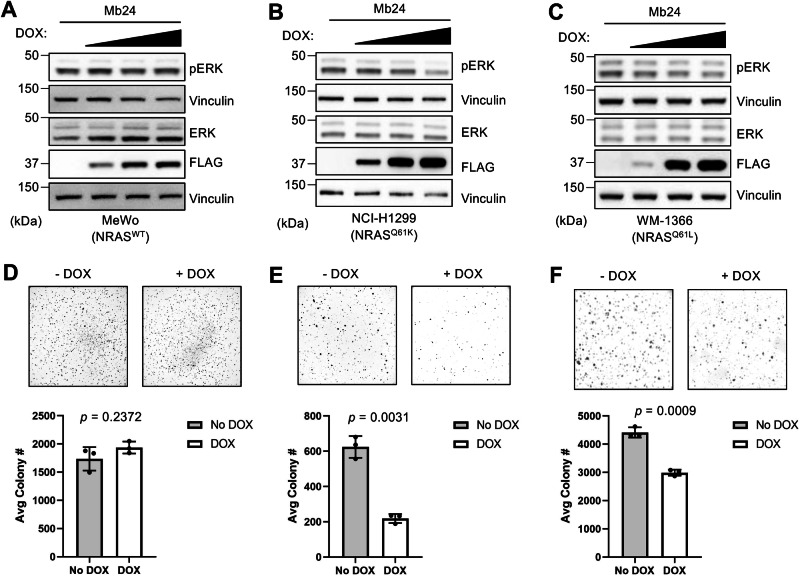
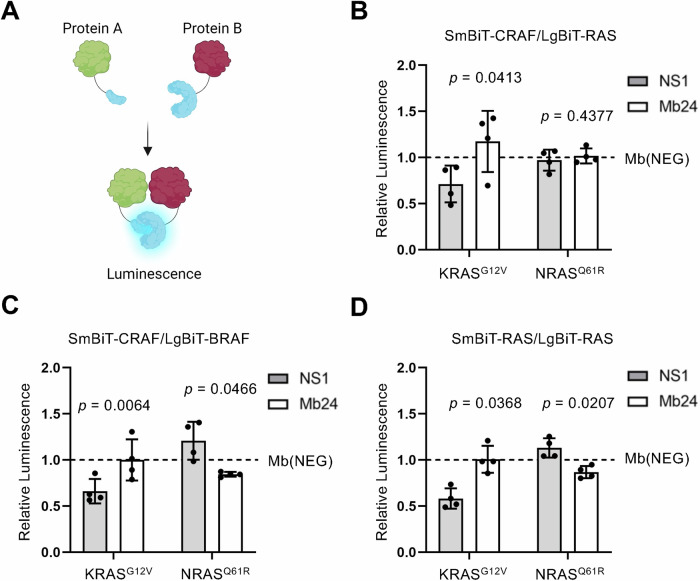
The RAS family GTPases are the most frequently mutated oncogene family in human cancers. Activating mutations in either of the three RAS isoforms (HRAS, KRAS, or NRAS) are found in nearly 20% of all human tumors with NRAS mutated in ~25% of melanomas. Despite remarkable advancements in therapies targeted against mutant KRAS, NRAS-specific pharmacologics are lacking. Thus, development of inhibitors of NRAS would address a critical unmet need to treating primary tumors harboring NRAS mutations as well as BRAF-mutant melanomas, which frequently develop resistance to clinically approved BRAF inhibitors through NRAS mutation. Building upon our previous studies with the monobody NS1 that recognizes HRAS and KRAS but not NRAS, here we report the development of a monobody that specifically binds to both GDP and GTP-bound states of NRAS and inhibits NRAS-mediated signaling in a mutation-agnostic manner. Further, this monobody can be formatted into a genetically encoded NRAS-specific degrader. Our study highlights the feasibility of developing NRAS selective inhibitors for therapeutic efforts.
© 2024. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
Competing interests: JPO, AK and SK are listed as inventors on a patent application on Monobodies targeting the nucleotide-free state of RAS files by the Medical University of South Carolina and New York University (No. 62/862,924). KWT, AK, and SK are listed as inventors on a patent application on mutant RAS targeting Monobodies filed by New York University (Application No. 63/121,903). AK and SK are listed as inventors on issued and pending patents on Monobody technology filed by The University of Chicago (US Patent 9512199 B2 and related pending applications), and on a pending patent on NRAS-selective monobodies (WO2023192915A1). SK is a co-founder and holds equity in Aethon Therapeutics; is a co-founder and holds equity in Revalia Bio; was an SAB member and received consulting fees from Black Diamond Therapeutics; has received research funding from Aethon Therapeutics, Argenx BVBA, Black Diamond Therapeutics, and Puretech Health. The other authors declare no competing interests. The other authors declare no competing interests. Ethics approval and consent to participate: This study did not use any human subjects, vertebrate animals or identifiable images from human research patients.
Figures
References
MeSH terms
Substances
Grants and funding
- GM130457/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- P30 CA016087/CA/NCI NIH HHS/United States
- BX002095/U.S. Department of Veterans Affairs (Department of Veterans Affairs)
- CA212608/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- P30 CA138313/CA/NCI NIH HHS/United States
- CA016087/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- GM132055/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- P20 GM130457/GM/NIGMS NIH HHS/United States
- CA138313/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R01 CA212608/CA/NCI NIH HHS/United States
- I01 BX002095/BX/BLRD VA/United States
- T32 GM132055/GM/NIGMS NIH HHS/United States
- CA194864/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R01 CA194864/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
