

Rare variant analyses in 51,256 type 2 diabetes cases and 370,487 controls reveal the pathogenicity spectrum of monogenic diabetes genes
- PMID: 39379762
- PMCID: PMC11549050
- DOI: 10.1038/s41588-024-01947-9
Rare variant analyses in 51,256 type 2 diabetes cases and 370,487 controls reveal the pathogenicity spectrum of monogenic diabetes genes
Erratum in
-
Publisher Correction: Rare variant analyses in 51,256 type 2 diabetes cases and 370,487 controls reveal the pathogenicity spectrum of monogenic diabetes genes.Nat Genet. 2024 Nov;56(11):2576. doi: 10.1038/s41588-024-01986-2. Nat Genet. 2024. PMID: 39438755 Free PMC article. No abstract available.
-
Author Correction: Rare variant analyses in 51,256 type 2 diabetes cases and 370,487 controls reveal the pathogenicity spectrum of monogenic diabetes genes.Nat Genet. 2025 Apr;57(4):1060. doi: 10.1038/s41588-025-02175-5. Nat Genet. 2025. PMID: 40133561 Free PMC article. No abstract available.
Abstract
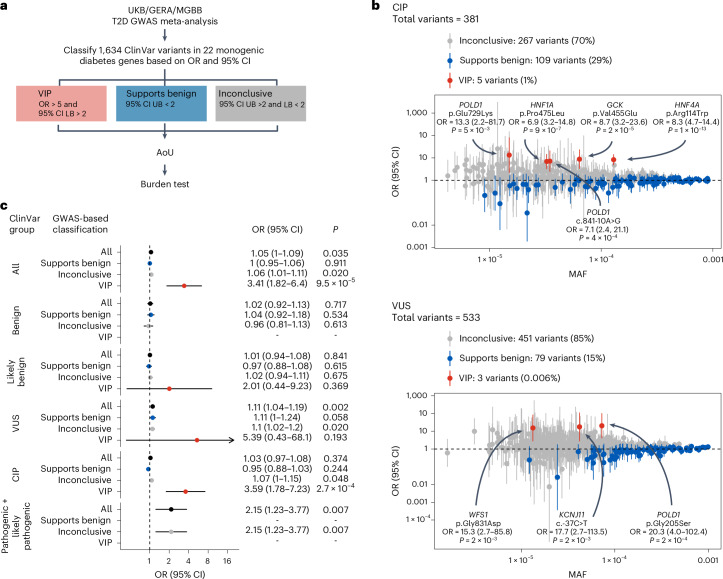
Type 2 diabetes (T2D) genome-wide association studies (GWASs) often overlook rare variants as a result of previous imputation panels' limitations and scarce whole-genome sequencing (WGS) data. We used TOPMed imputation and WGS to conduct the largest T2D GWAS meta-analysis involving 51,256 cases of T2D and 370,487 controls, targeting variants with a minor allele frequency as low as 5 × 10-5. We identified 12 new variants, including a rare African/African American-enriched enhancer variant near the LEP gene (rs147287548), associated with fourfold increased T2D risk. We also identified a rare missense variant in HNF4A (p.Arg114Trp), associated with eightfold increased T2D risk, previously reported in maturity-onset diabetes of the young with reduced penetrance, but observed here in a T2D GWAS. We further leveraged these data to analyze 1,634 ClinVar variants in 22 genes related to monogenic diabetes, identifying two additional rare variants in HNF1A and GCK associated with fivefold and eightfold increased T2D risk, respectively, the effects of which were modified by the individual's polygenic risk score. For 21% of the variants with conflicting interpretations or uncertain significance in ClinVar, we provided support of being benign based on their lack of association with T2D. Our work provides a framework for using rare variant GWASs to identify large-effect variants and assess variant pathogenicity in monogenic diabetes genes.
© 2024. The Author(s).
Conflict of interest statement
A.L.G.’s spouse is employed by Genentech and holds stock options in Roche. A.K.M. is an unpaid research collaborator with AstraZeneca. J.M.M. has research funded in collaboration with Novo Nordisk. The other authors declare no competing interests.
Figures














Update of
-
Rare variant association analysis in 51,256 type 2 diabetes cases and 370,487 controls informs the spectrum of pathogenicity of monogenic diabetes genes.medRxiv [Preprint]. 2023 Sep 29:2023.09.28.23296244. doi: 10.1101/2023.09.28.23296244. medRxiv. 2023. Update in: Nat Genet. 2024 Nov;56(11):2370-2379. doi: 10.1038/s41588-024-01947-9. PMID: 37808701 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
- OT2 OD026556/OD/NIH HHS/United States
- U2C OD023196/OD/NIH HHS/United States
- OT2 OD025315/OD/NIH HHS/United States
- OT2 OD026551/OD/NIH HHS/United States
- U24 OD023121/OD/NIH HHS/United States
- OT2 OD026552/OD/NIH HHS/United States
- OT2 OD026549/OD/NIH HHS/United States
- OT2 OD025337/OD/NIH HHS/United States
- OT2 OD026555/OD/NIH HHS/United States
- OT2 OD026550/OD/NIH HHS/United States
- U01 HG011723/HG/NHGRI NIH HHS/United States
- OT2 OD026553/OD/NIH HHS/United States
- OT2 OD023205/OD/NIH HHS/United States
- OT2 OD025276/OD/NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- K23 DK131345/DK/NIDDK NIH HHS/United States
- OT2 OD023206/OD/NIH HHS/United States
- U24 OD023176/OD/NIH HHS/United States
- OT2 OD026548/OD/NIH HHS/United States
- OT2 OD025277/OD/NIH HHS/United States
- U01HG011723/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- OT2 OD026557/OD/NIH HHS/United States
- UM1 DK078616/DK/NIDDK NIH HHS/United States
- OT2 OD026554/OD/NIH HHS/United States
- R01 DK140555/DK/NIDDK NIH HHS/United States
- K24 HL157960/HL/NHLBI NIH HHS/United States
- UM1 DK126185/DK/NIDDK NIH HHS/United States
- U24 OD023163/OD/NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
