

Tumor Integrin-Targeted Glucose Oxidase Enzyme Promotes ROS-Mediated Cell Death that Combines with Interferon Alpha Therapy for Tumor Control
- PMID: 39382078
- PMCID: PMC11695183
- DOI: 10.1158/1535-7163.MCT-24-0163
Tumor Integrin-Targeted Glucose Oxidase Enzyme Promotes ROS-Mediated Cell Death that Combines with Interferon Alpha Therapy for Tumor Control
Abstract
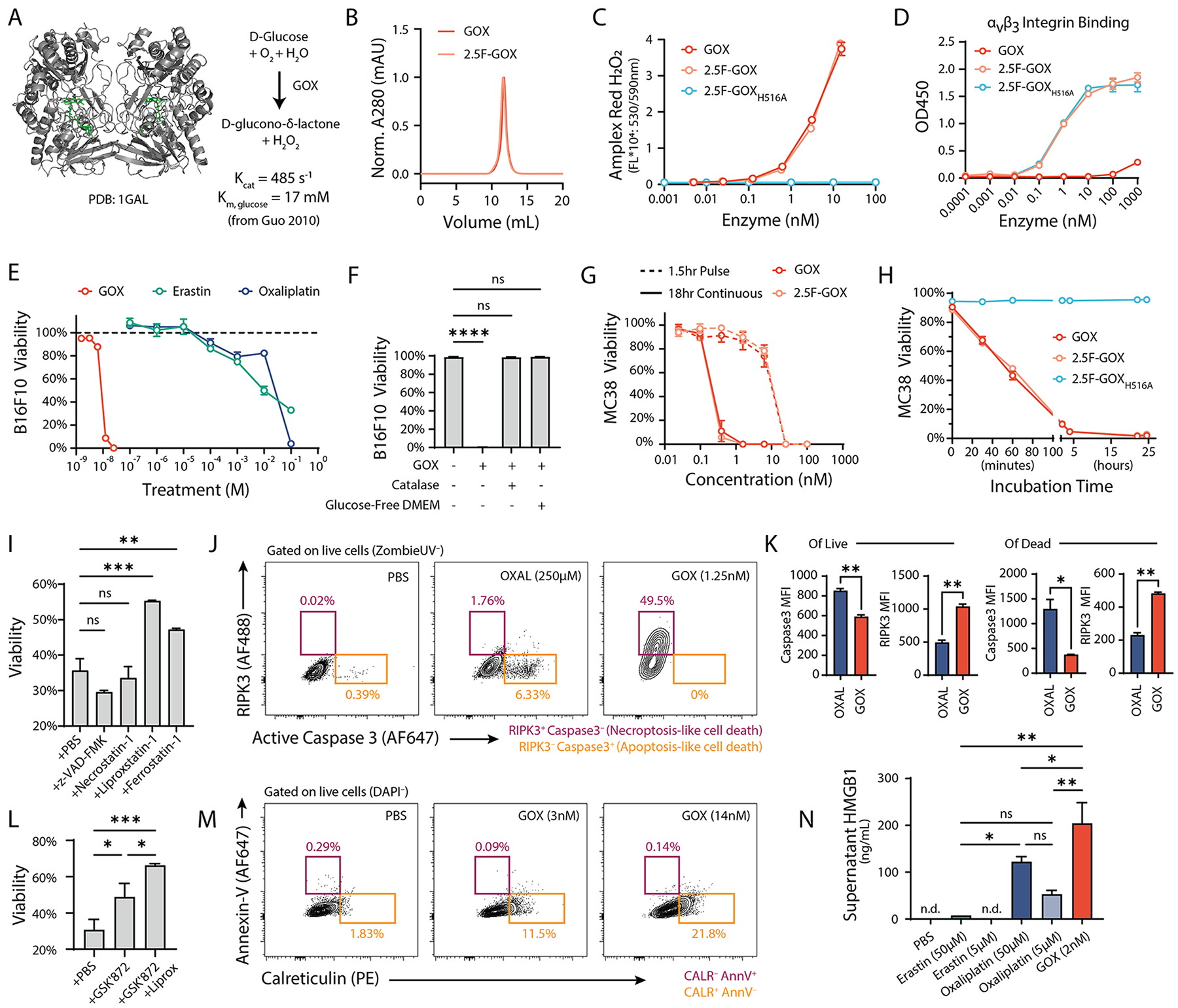
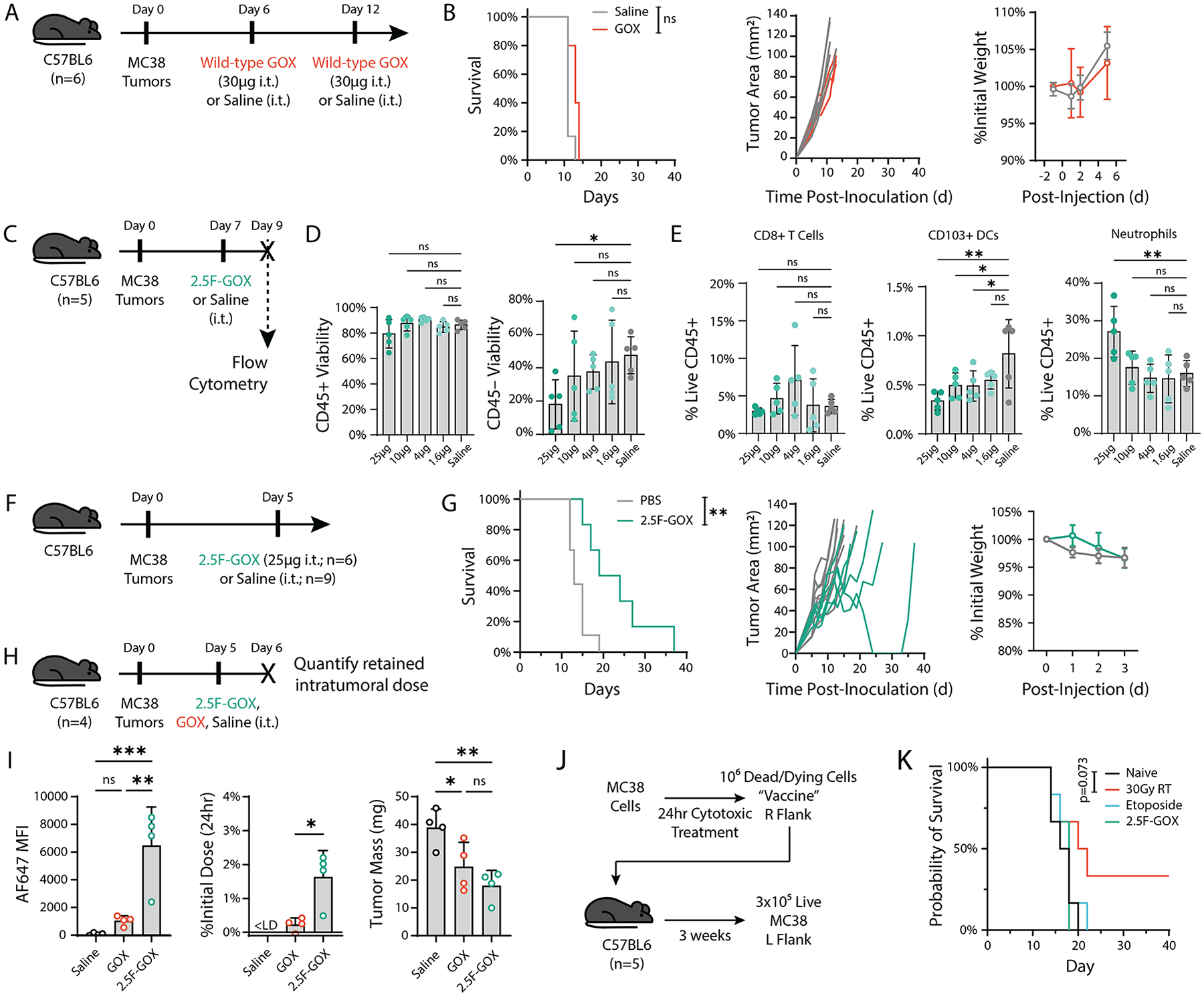
Although heightened intratumoral levels of reactive oxygen species (ROS) are typically associated with a suppressive tumor microenvironment, under certain conditions ROS contribute to tumor elimination. Treatment approaches, including some chemotherapy and radiation protocols, increase cancer cell ROS levels that influence their mechanism of cell death and subsequent recognition by the immune system. Furthermore, activated myeloid cells rapidly generate ROS upon encounter with pathogens or infected cells to eliminate disease, and recently, this effector function has been noted in cancer contexts as well. Collectively, ROS-induced cancer cell death may help initiate adaptive antitumor immune responses that could synergize with current approved immunotherapies, for improved control of solid tumors. In this work, we explore the use of glucose oxidase, an enzyme which produces hydrogen peroxide, a type of ROS, to therapeutically mimic the endogenous oxidative burst from myeloid cells to promote antigen generation within the tumor microenvironment. We engineer the enzyme to target pan-tumor-expressed integrins both as a tumor-agnostic therapeutic approach and as a strategy to prolong local enzyme activity following intratumoral administration. We found the targeted enzyme potently induced cancer cell death and enhanced cross-presentation by dendritic cells in vitro and further combined with interferon alpha for long-term tumor control in murine MC38 tumors in vivo. Optimizing the single-dose administration of this enzyme overcomes limitations with immunogenicity noted for other prooxidant enzyme approaches. Overall, our results suggest ROS-induced cell death can be harnessed for tumor control and highlight the potential use of designed enzyme therapies alongside immunotherapy against cancer.
©2024 American Association for Cancer Research.
Conflict of interest statement
CONFLICT OF INTEREST DISCLOSURE STATEMENT
The authors declare no potential conflicts of interest.
Figures


Similar articles
-
Immunotherapy for metastatic renal cell carcinoma.Cochrane Database Syst Rev. 2017 May 15;5(5):CD011673. doi: 10.1002/14651858.CD011673.pub2. Cochrane Database Syst Rev. 2017. PMID: 28504837 Free PMC article.
-
Cost-effectiveness of using prognostic information to select women with breast cancer for adjuvant systemic therapy.Health Technol Assess. 2006 Sep;10(34):iii-iv, ix-xi, 1-204. doi: 10.3310/hta10340. Health Technol Assess. 2006. PMID: 16959170
-
A rapid and systematic review of the clinical effectiveness and cost-effectiveness of paclitaxel, docetaxel, gemcitabine and vinorelbine in non-small-cell lung cancer.Health Technol Assess. 2001;5(32):1-195. doi: 10.3310/hta5320. Health Technol Assess. 2001. PMID: 12065068
-
EORTC guidelines for the use of erythropoietic proteins in anaemic patients with cancer: 2006 update.Eur J Cancer. 2007 Jan;43(2):258-70. doi: 10.1016/j.ejca.2006.10.014. Epub 2006 Dec 19. Eur J Cancer. 2007. PMID: 17182241
-
Thrombolysis for acute ischaemic stroke.Cochrane Database Syst Rev. 2003;(3):CD000213. doi: 10.1002/14651858.CD000213. Cochrane Database Syst Rev. 2003. Update in: Cochrane Database Syst Rev. 2009 Oct 07;(4):CD000213. doi: 10.1002/14651858.CD000213.pub2. PMID: 12917889 Updated.
References
-
- Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–3. - PubMed
-
- Finkel T. Oxygen radicals and signaling. Curr Opin Cell Biol. 1998;10:248–53. - PubMed
-
- Rhee SG, Bae YS, Lee SR, Kwon J. Hydrogen peroxide: a key messenger that modulates protein phosphorylation through cysteine oxidation. Sci STKE. 2000;2000:e1. - PubMed
-
- Miki H, Funato Y. Regulation of intracellular signalling through cysteine oxidation by reactive oxygen species. J Biochem. 2012;151:255–61. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
