

Histone modifications of circulating nucleosomes are associated with changes in cell-free DNA fragmentation patterns
- PMID: 39382996
- PMCID: PMC11494292
- DOI: 10.1073/pnas.2404058121
Histone modifications of circulating nucleosomes are associated with changes in cell-free DNA fragmentation patterns
Abstract
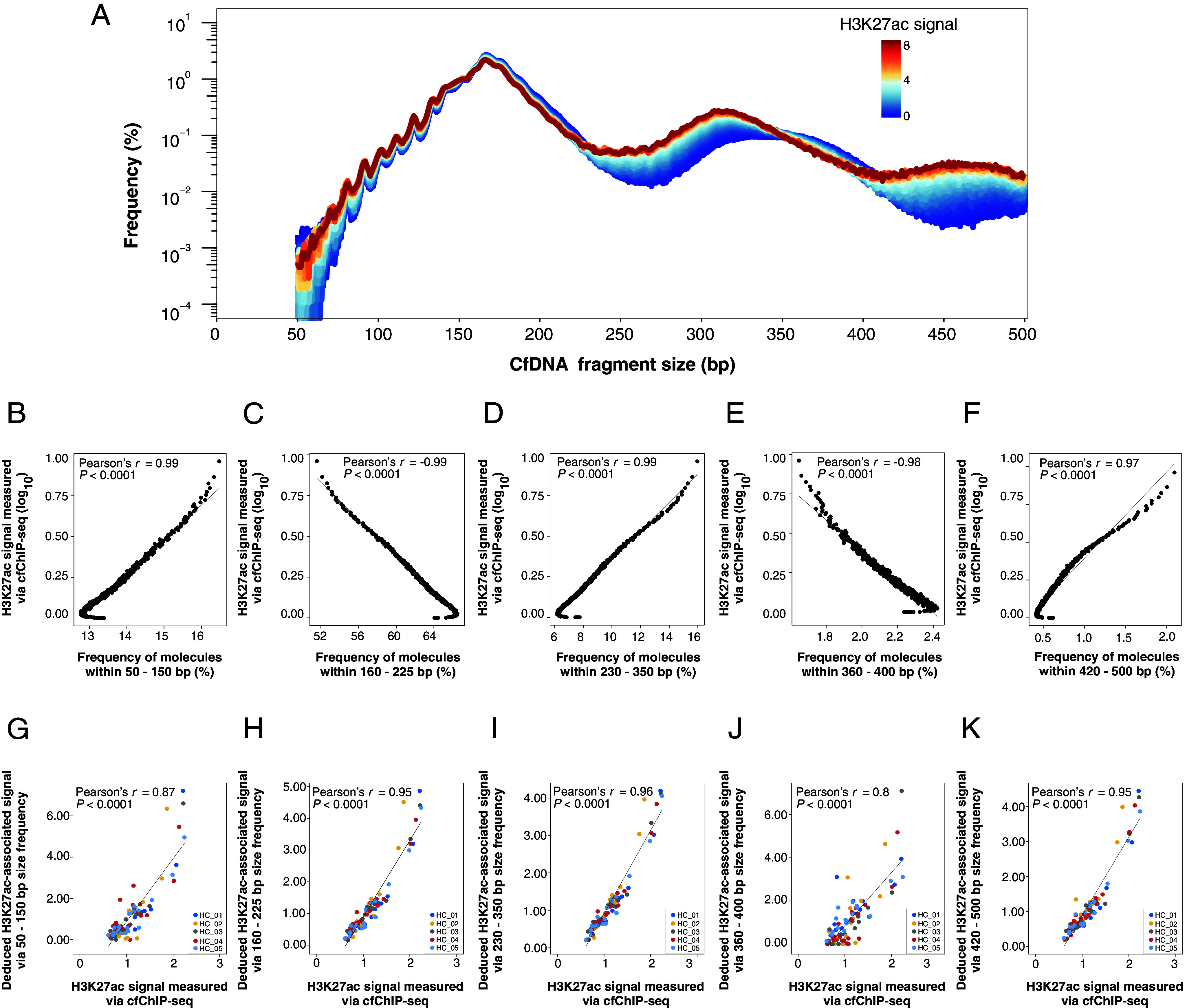
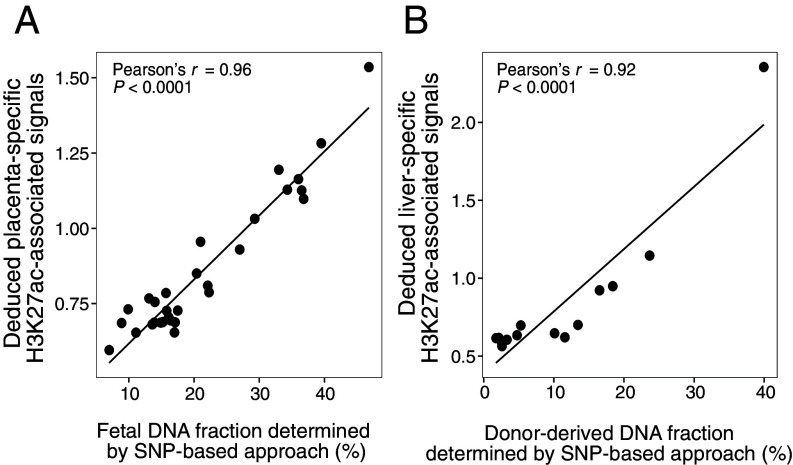
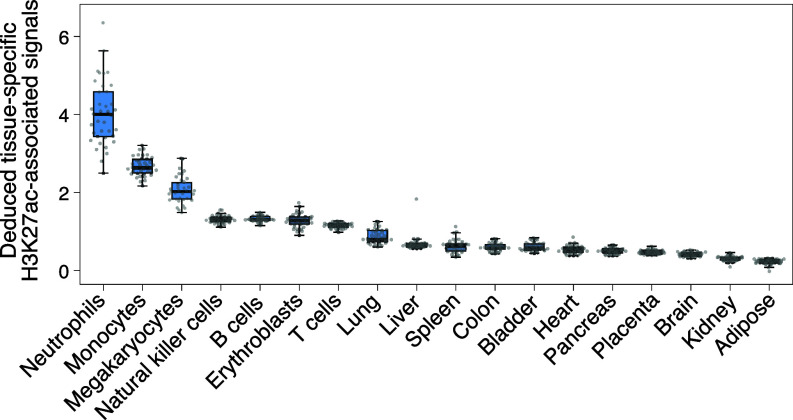
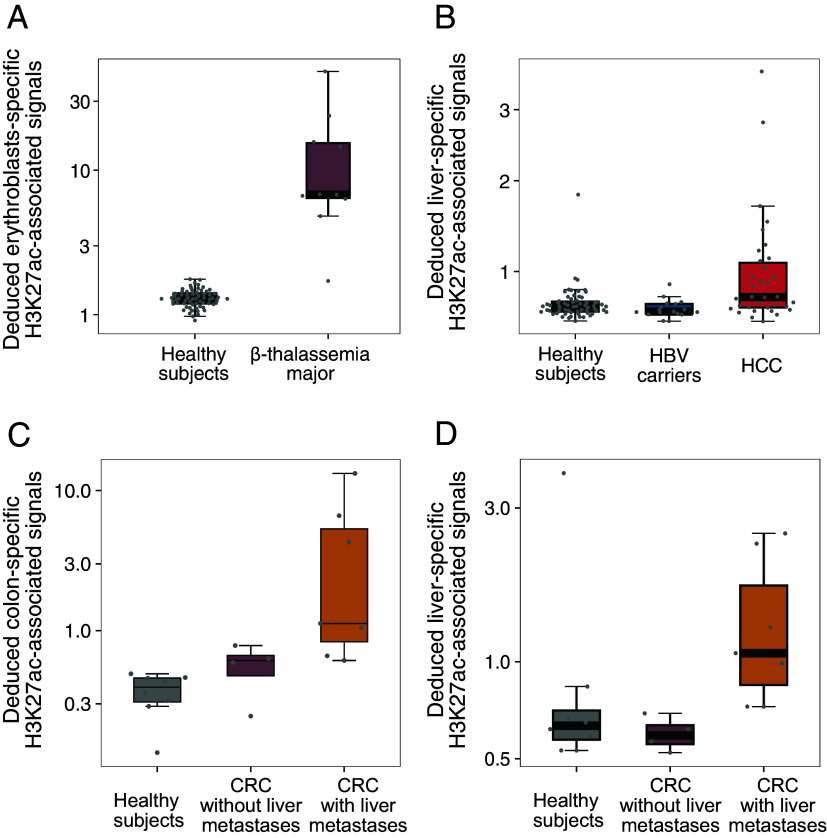
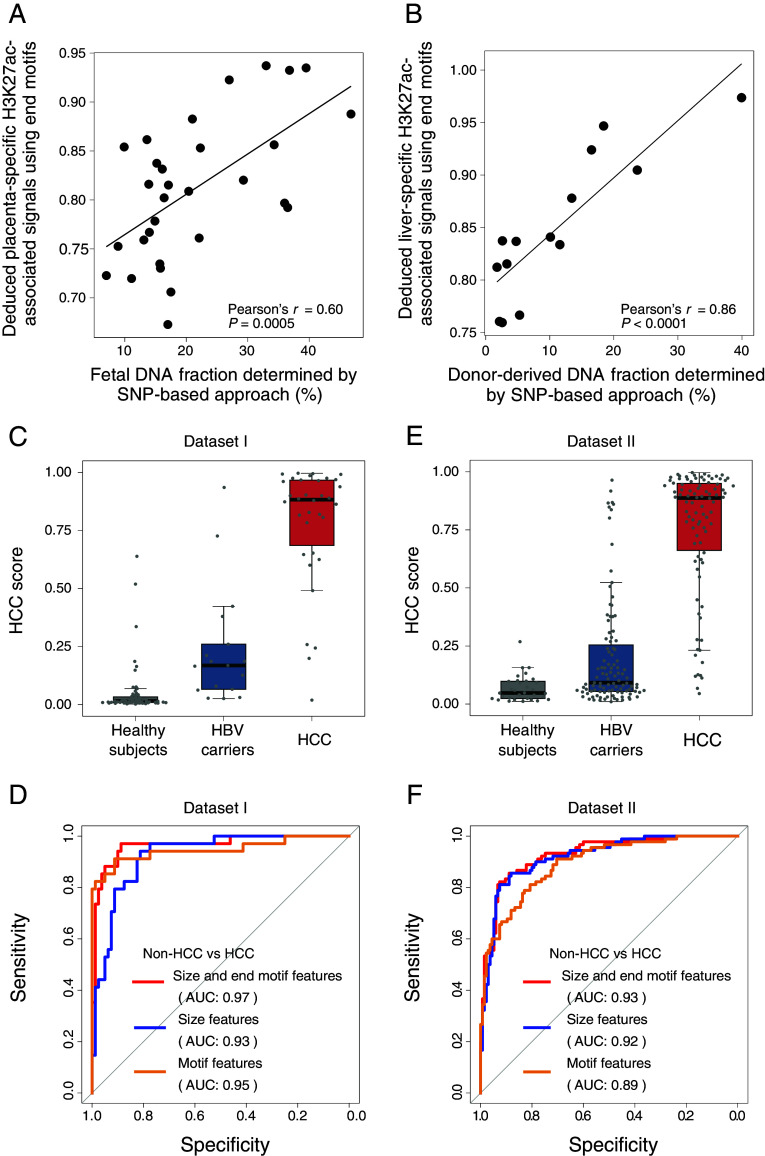
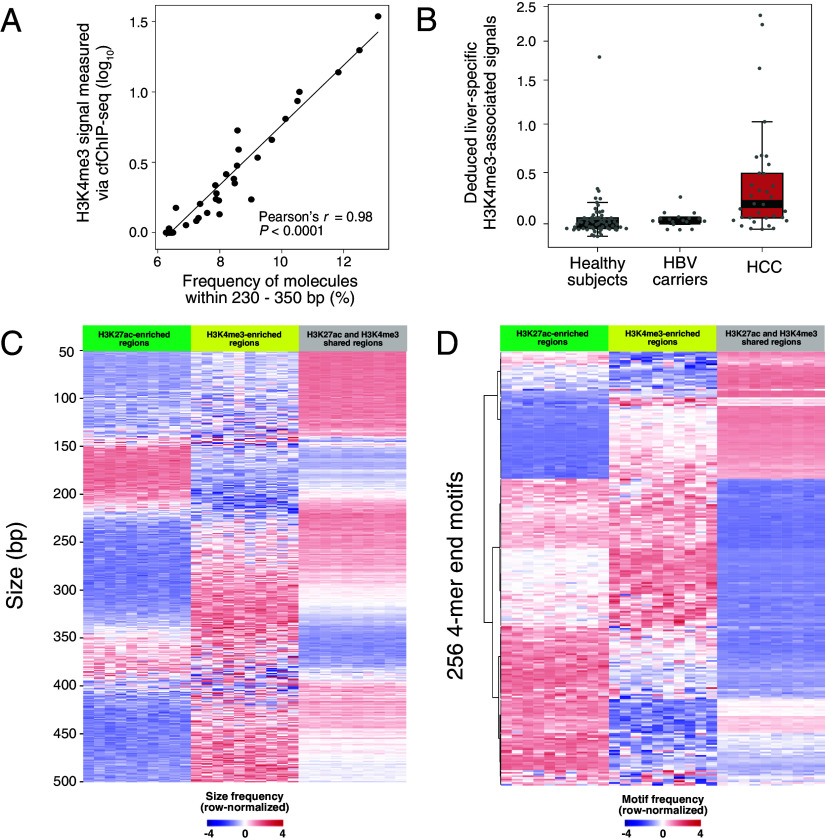
The analysis of tissues of origin of cell-free DNA (cfDNA) is of research and diagnostic interest. Many studies focused on bisulfite treatment or immunoprecipitation protocols to assess the tissues of origin of cfDNA. DNA loss often occurs during such processes. Fragmentomics of cfDNA molecules has uncovered a wealth of information related to tissues of origin of cfDNA. There is still much room for the development of tools for assessing contributions from various tissues into plasma using fragmentomic features. Hence, we developed an approach to analyze the relative contributions of DNA from different tissues into plasma, by identifying characteristic fragmentation patterns associated with selected histone modifications. We named this technique as FRAGmentomics-based Histone modification Analysis (FRAGHA). Deduced placenta-specific histone H3 lysine 27 acetylation (H3K27ac)-associated signal correlated well with the fetal DNA fraction in maternal plasma (Pearson's r = 0.96). The deduced liver-specific H3K27ac-associated signal correlated with the donor-derived DNA fraction in liver transplantation recipients (Pearson's r = 0.92) and was significantly increased in patients with hepatocellular carcinoma (HCC) (P < 0.01, Wilcoxon rank-sum test). Significant elevations of erythroblasts-specific and colon-specific H3K27ac-associated signals were observed in patients with β-thalassemia major and colorectal cancer, respectively. Furthermore, using the fragmentation patterns from tissue-specific H3K27ac regions, a machine learning algorithm was developed to enhance HCC detection, with an area under the curve (AUC) of up to 0.97. Finally, genomic regions with H3K27ac or histone H3 lysine 4 trimethylation (H3K4me3) were found to exhibit different fragmentomic patterns of cfDNA. This study has shed light on the relationship between cfDNA fragmentomics and histone modifications, thus expanding the armamentarium of liquid biopsy.
Keywords: epigenetics; fragmentomics; histone modifications; liquid biopsy.
Conflict of interest statement
Competing interests statement:J.B., P.J., L.J., M.Y., K.C.A.C., and Y.M.D.L. filed patent applications based on the data in this study. Reviewer S.B. is an inventor on patents related to cfDNA mutation and methylation analysis technologies that have been licensed to Roche and Adela, respectively, and is a co-founder and has ownership in Adela.
Figures
References
-
- Lo Y. M. D., Han D. S. C., Jiang P., Chiu R. W. K., Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science 372, eaaw3616 (2021). - PubMed
-
- Rostami A., et al. , Senescence, necrosis, and apoptosis govern circulating cell-free DNA release kinetics. Cell Rep. 31, 107830 (2020). - PubMed
-
- Lo Y. M., et al. , Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Transl. Med. 2, 61ra91 (2010). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
