

Steering research on mRNA splicing in cancer towards clinical translation
- PMID: 39384951
- PMCID: PMC11698124
- DOI: 10.1038/s41568-024-00750-2
Steering research on mRNA splicing in cancer towards clinical translation
Abstract
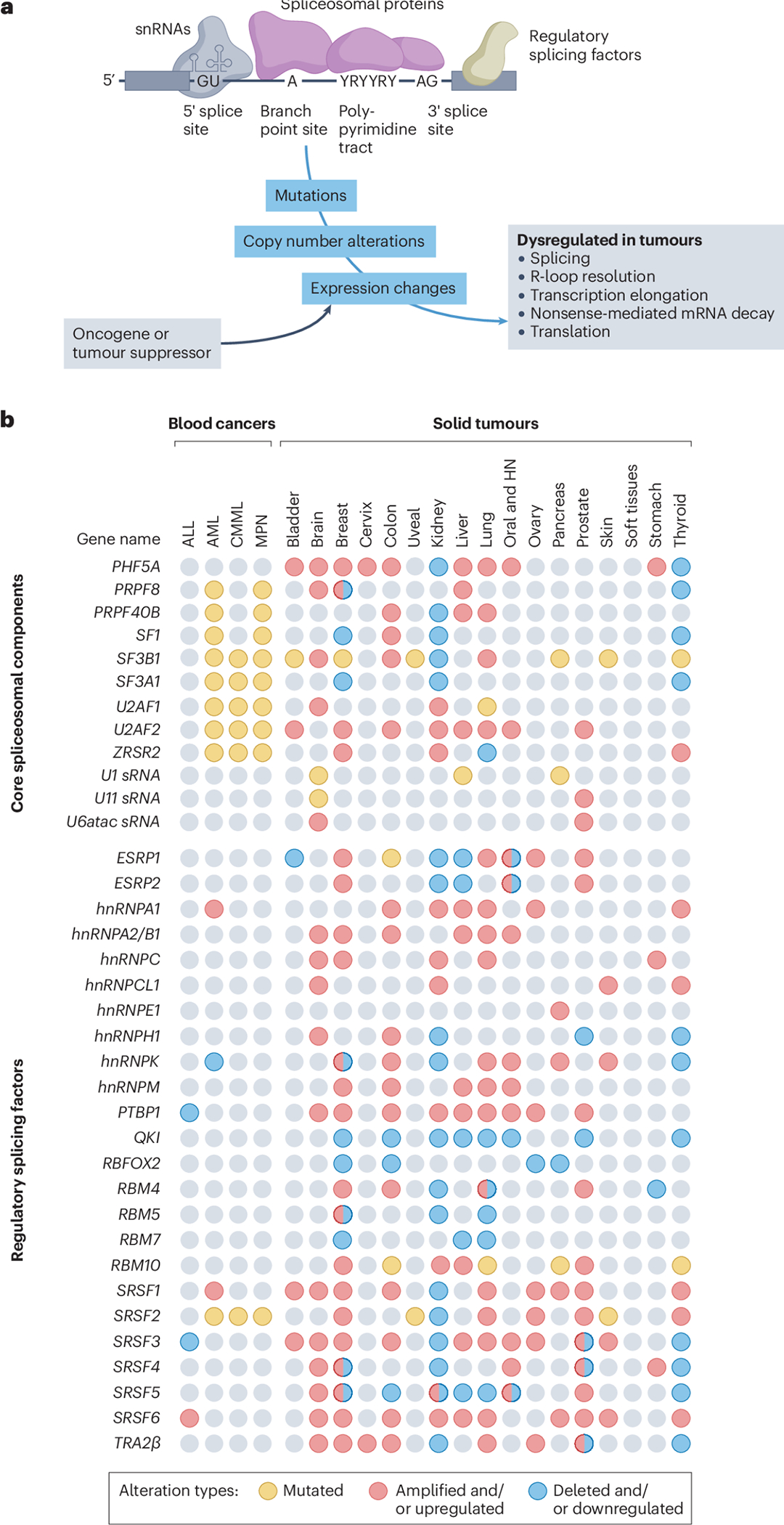
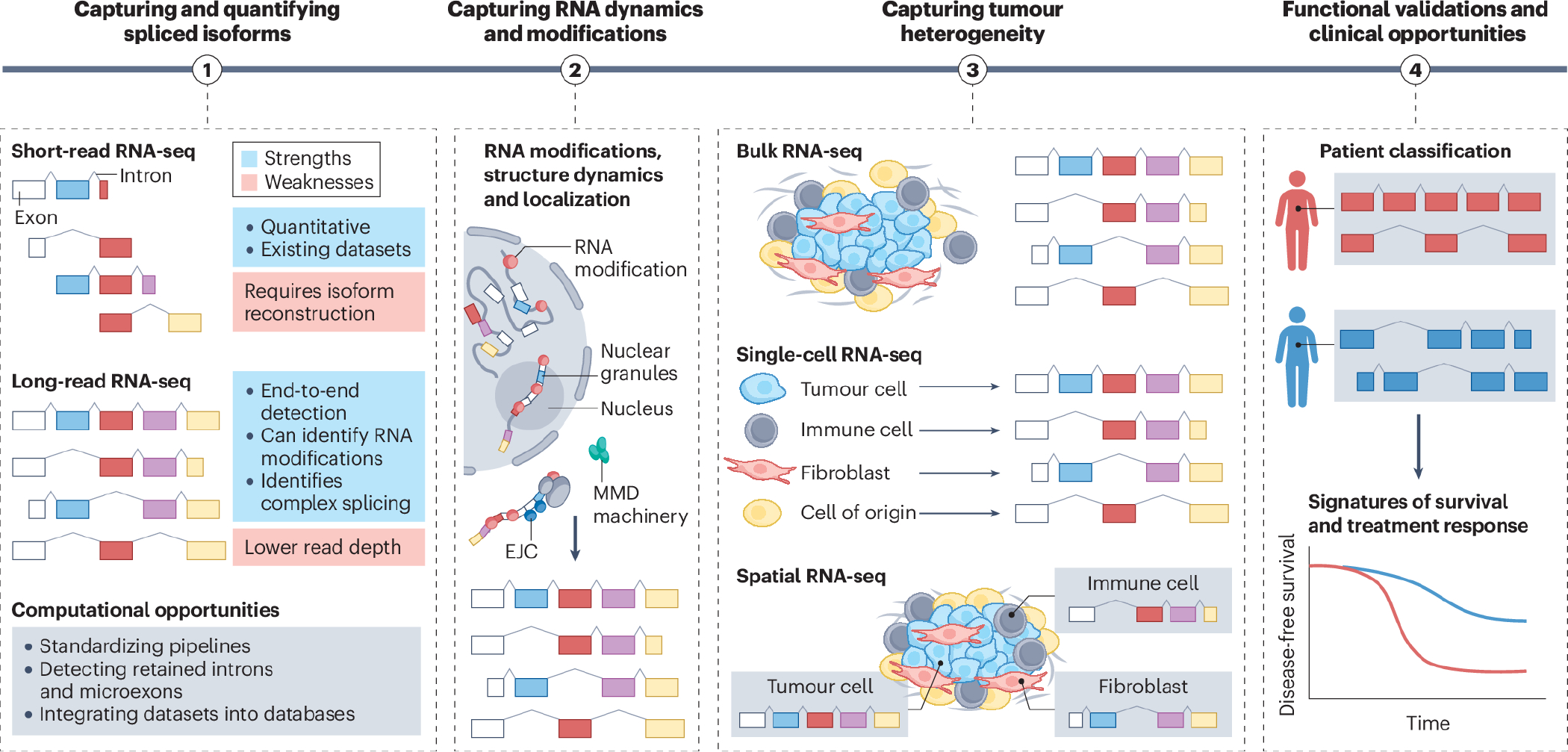
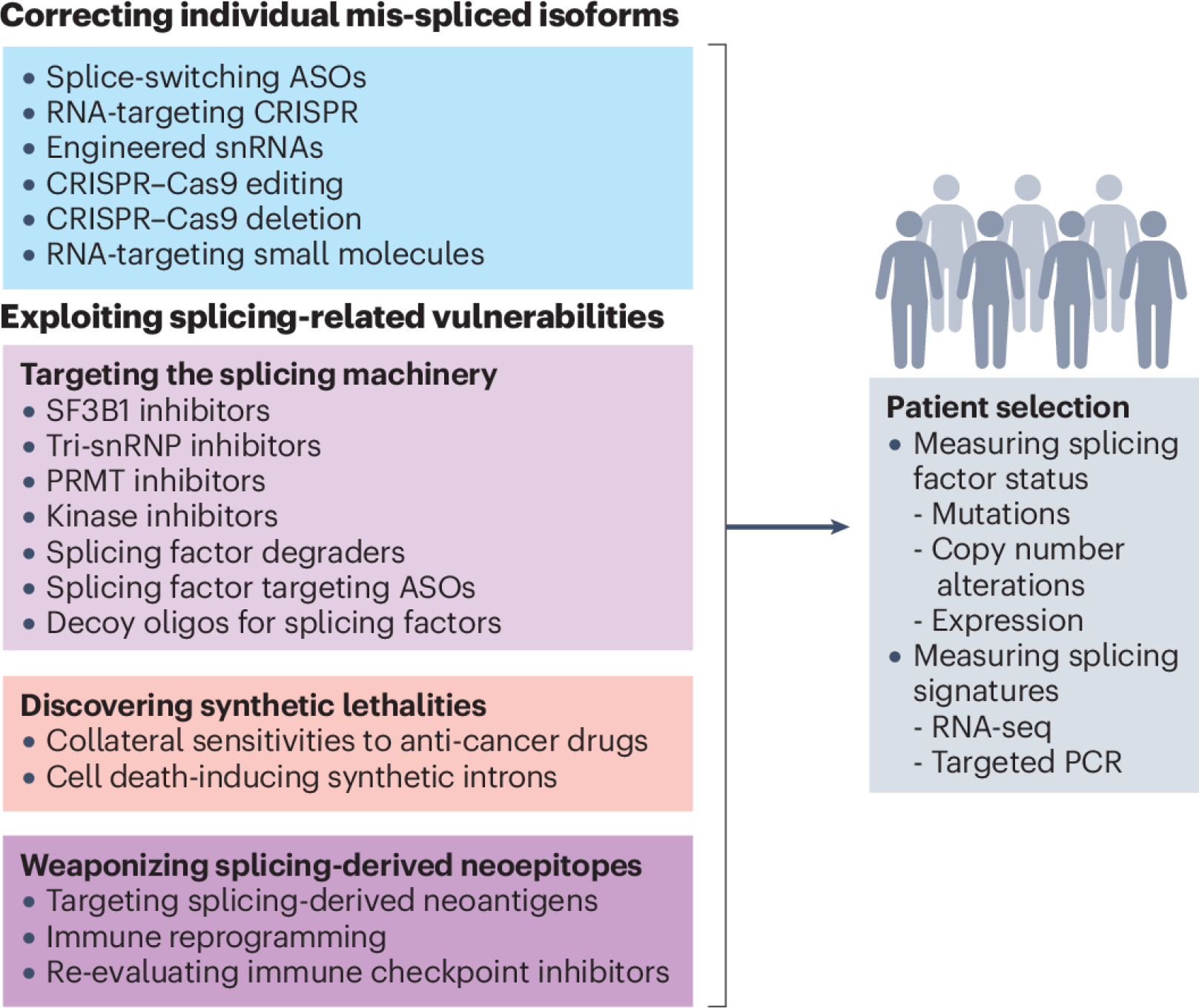
Splicing factors are affected by recurrent somatic mutations and copy number variations in several types of haematologic and solid malignancies, which is often seen as prima facie evidence that splicing aberrations can drive cancer initiation and progression. However, numerous spliceosome components also 'moonlight' in DNA repair and other cellular processes, making their precise role in cancer difficult to pinpoint. Still, few would deny that dysregulated mRNA splicing is a pervasive feature of most cancers. Correctly interpreting these molecular fingerprints can reveal novel tumour vulnerabilities and untapped therapeutic opportunities. Yet multiple technological challenges, lingering misconceptions, and outstanding questions hinder clinical translation. To start with, the general landscape of splicing aberrations in cancer is not well defined, due to limitations of short-read RNA sequencing not adept at resolving complete mRNA isoforms, as well as the shallow read depth inherent in long-read RNA-sequencing, especially at single-cell level. Although individual cancer-associated isoforms are known to contribute to cancer progression, widespread splicing alterations could be an equally important and, perhaps, more readily actionable feature of human cancers. This is to say that in addition to 'repairing' mis-spliced transcripts, possible therapeutic avenues include exacerbating splicing aberration with small-molecule spliceosome inhibitors, targeting recurrent splicing aberrations with synthetic lethal approaches, and training the immune system to recognize splicing-derived neoantigens.
© 2024. Springer Nature Limited.
Conflict of interest statement
Competing interests: J.V. is a member of the Advisory Boards of Remix Therapeutics, Stoke Therapeutics and IntronX. K.M.W. is an adviser to and holds equity in Ribometrix, ForagR Medicines and A-Form Solutions. O.A. is a member of the Advisory Boards of Caeruleus Genomics.
Figures
References
-
- Chow LT, Gelinas RE, Broker TR & Roberts RJ An amazing sequence arrangement at the 5’ ends of adenovirus 2 messenger RNA. Cell 12, 1–8 (1977). - PubMed
-
- Pan Q, Shai O, Lee LJ, Frey BJ & Blencowe BJ Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40, 1413–1415 (2008). - PubMed
-
- Kjer-Hansen P & Weatheritt RJ The function of alternative splicing in the proteome: rewiring protein interactomes to put old functions into new contexts. Nat Struct Mol Biol 30, 1844–1856 (2023). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- K08 CA245242/CA/NCI NIH HHS/United States
- R01 CA249204/CA/NCI NIH HHS/United States
- U01 CA232563/CA/NCI NIH HHS/United States
- R01 HL167071/HL/NHLBI NIH HHS/United States
- P30 CA034196/CA/NCI NIH HHS/United States
- R01 CA248317/CA/NCI NIH HHS/United States
- R35 GM131876/GM/NIGMS NIH HHS/United States
- R35 GM136426/GM/NIGMS NIH HHS/United States
- R01 GM138541/GM/NIGMS NIH HHS/United States
- R01 GM140735/GM/NIGMS NIH HHS/United States
- P30 CA196521/CA/NCI NIH HHS/United States
- R21 AG080243/AG/NIA NIH HHS/United States
- R01 CA182467/CA/NCI NIH HHS/United States
