

AKT and EZH2 inhibitors kill TNBCs by hijacking mechanisms of involution
- PMID: 39385030
- PMCID: PMC11578877
- DOI: 10.1038/s41586-024-08031-6
AKT and EZH2 inhibitors kill TNBCs by hijacking mechanisms of involution
Abstract
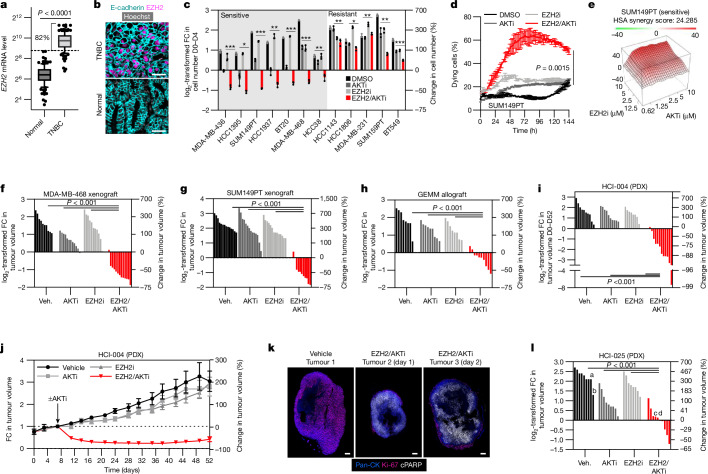
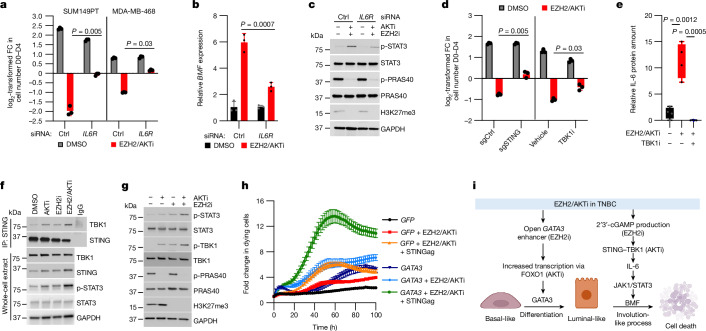
Triple-negative breast cancer (TNBC) is the most aggressive breast cancer subtype and has the highest rate of recurrence1. The predominant standard of care for advanced TNBC is systemic chemotherapy with or without immunotherapy; however, responses are typically short lived1,2. Thus, there is an urgent need to develop more effective treatments. Components of the PI3K pathway represent plausible therapeutic targets; more than 70% of TNBCs have alterations in PIK3CA, AKT1 or PTEN3-6. However, in contrast to hormone-receptor-positive tumours, it is still unclear whether or how triple-negative disease will respond to PI3K pathway inhibitors7. Here we describe a promising AKT-inhibitor-based therapeutic combination for TNBC. Specifically, we show that AKT inhibitors synergize with agents that suppress the histone methyltransferase EZH2 and promote robust tumour regression in multiple TNBC models in vivo. AKT and EZH2 inhibitors exert these effects by first cooperatively driving basal-like TNBC cells into a more differentiated, luminal-like state, which cannot be effectively induced by either agent alone. Once TNBCs are differentiated, these agents kill them by hijacking signals that normally drive mammary gland involution. Using a machine learning approach, we developed a classifier that can be used to predict sensitivity. Together, these findings identify a promising therapeutic strategy for this highly aggressive tumour type and illustrate how deregulated epigenetic enzymes can insulate tumours from oncogenic vulnerabilities. These studies also reveal how developmental tissue-specific cell death pathways may be co-opted for therapeutic benefit.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: K.C. is an advisor at Genentech and serves on the scientific advisory board of Erasca. S.M.T. has consulting or advisory roles at Novartis, Pfizer, Merck, Lilly, Nektar, NanoString Technologies, AstraZeneca, Puma Biotechnology, Genentech/Roche, Eisai, Sanofi, Bristol Myers Squibb, Seattle Genetics, Odonate Therapeutics, OncoPep, Kyowa Hakko Kirin, Samsung Bioepis, CytomX Therapeutics, Daiichi Sankyo, Athenex, Gilead, Mersana, Certara, Chugai Pharma, Ellipses Pharma, Infinity, 4D Pharma, OncoSec Medical, BeyondSpring Pharmaceuticals, OncXerna, Zymeworks, Zentalis, Blueprint Medicines, Reveal Genomics and ARC Therapeutics; and received institutional research funding from Genentech/Roche, Merck, Exelixis, Pfizer, Lilly, Novartis, Bristol Myers Squibb, Eisai, AstraZeneca, NanoString Technologies, Cyclacel, Nektar, Gilead, Odonate Therapeutics, Sanofi and Seattle Genetics. P.K.S. is a co-founder and member of the BOD of Glencoe Software, member of the BOD for Applied Biomath, member of the scientific advisory board for RareCyte, NanoString and Montai Health, holds equity in Glencoe, Applied Biomath and RareCyte, consults for Merck, and has received research funding to the institution from Novartis and Merck in the past 5 years. A.C.G.-C. has grant/research funding to institution from Gilead Sciences, AstraZeneca, Daiichi-Sankyo, Merck, Zenith Epigenetics, Bristol-Myers Squibb, Novartis, Foundation Medicine and Biovica; serves as a consultant and member of the scientific advisory board for AstraZeneca, Daiichi-Sankyo and Novartis; has received honoraria from AstraZeneca and Daiichi-Sankyo; and has other financial or materials support from Roche/Genentech, Gilead Sciences, AstraZeneca, Novartis and Merck. K.H. is a consultant for and a co-founder of Dania Therapeutics Aps and a scientific advisor for Hannibal Health Innovation. S.R.V.K. is a founder and consultant at Faeth Therapeutics and Transomic Technologies. D.A.B. is a consultant for N of One/QIAGEN and Tango Therapeutics; is a founder and shareholder in Xsphera Biosciences; has received honoraria from Merck, H3 Biomedicine/Esai, EMD Serono, Gilead Sciences, Abbvie and Madalon Consulting; and has received research grants from BMS, Takeda, Novartis, Gilead and Lilly. The other authors declare no competing interests.
Figures














References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
