

AAV gene therapy for Duchenne muscular dystrophy: the EMBARK phase 3 randomized trial
- PMID: 39385046
- PMCID: PMC11750718
- DOI: 10.1038/s41591-024-03304-z
AAV gene therapy for Duchenne muscular dystrophy: the EMBARK phase 3 randomized trial
Abstract
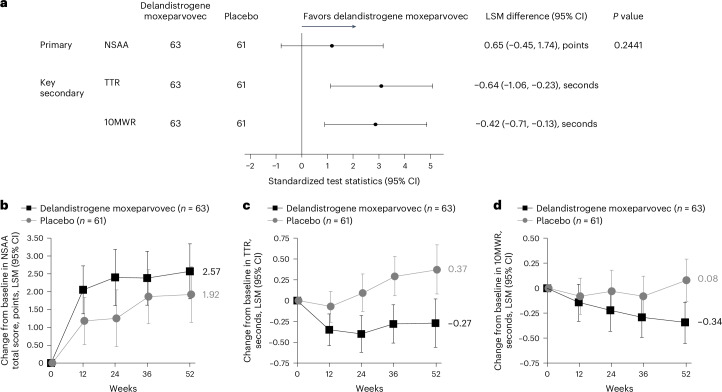
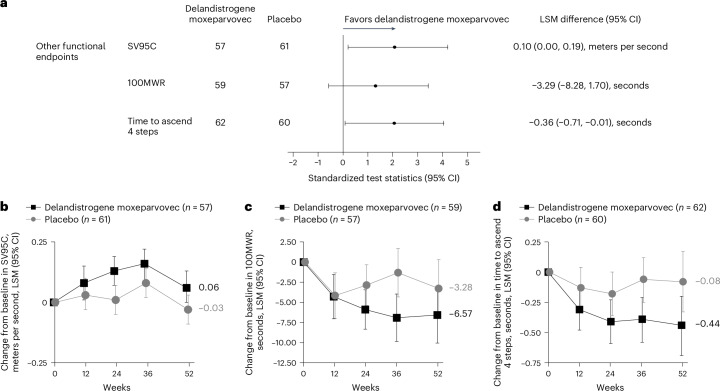
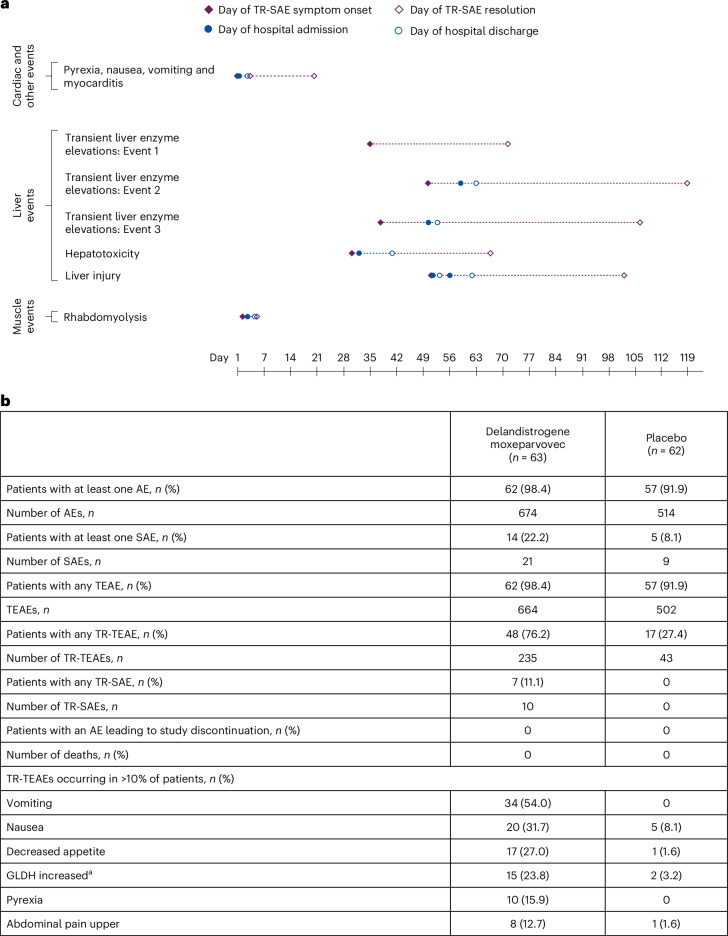
Duchenne muscular dystrophy (DMD) is a rare, X-linked neuromuscular disease caused by pathogenic variants in the DMD gene that result in the absence of functional dystrophin, beginning at birth and leading to progressive impaired motor function, loss of ambulation and life-threatening cardiorespiratory complications. Delandistrogene moxeparvovec, an adeno-associated rh74-viral vector-based gene therapy, addresses absent functional dystrophin in DMD. Here the phase 3 EMBARK study aimed to assess the efficacy and safety of delandistrogene moxeparvovec in patients with DMD. Ambulatory males with DMD, ≥4 years to <8 years of age, were randomized and stratified by age group and North Star Ambulatory Assessment (NSAA) score to single-administration intravenous delandistrogene moxeparvovec (1.33 × 1014 vector genomes per kilogram; n = 63) or placebo (n = 62). At week 52, the primary endpoint, change from baseline in NSAA score, was not met (least squares mean 2.57 (delandistrogene moxeparvovec) versus 1.92 (placebo) points; between-group difference, 0.65; 95% confidence interval (CI), -0.45, 1.74; P = 0.2441). Secondary efficacy endpoints included mean micro-dystrophin expression at week 12: 34.29% (treated) versus 0.00% (placebo). Other secondary efficacy endpoints at week 52 (between-group differences (95% CI)) included: Time to Rise (-0.64 (-1.06, -0.23)), 10-meter Walk/Run (-0.42 (-0.71, -0.13)), stride velocity 95th centile (0.10 (0.00, 0.19)), 100-meter Walk/Run (-3.29 (-8.28, 1.70)), time to ascend 4 steps (-0.36 (-0.71, -0.01)), PROMIS Mobility and Upper Extremity (0.05 (-0.08, 0.19); -0.04 (-0.24, 0.17)) and number of NSAA skills gained/improved (0.19 (-0.67, 1.06)). In total, 674 adverse events were recorded with delandistrogene moxeparvovec and 514 with placebo. There were no deaths, discontinuations or clinically significant complement-mediated adverse events; 7 patients (11.1%) experienced 10 treatment-related serious adverse events. Delandistrogene moxeparvovec did not lead to a significant improvement in NSAA score at week 52. Some of the secondary endpoints numerically favored treatment, although no statistical significance can be claimed. Safety was manageable and consistent with previous delandistrogene moxeparvovec trials. ClinicalTrials.gov: NCT05096221.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: J.R.M. received study funding from Sarepta Therapeutics while at Nationwide Children’s Hospital at the time of the study and is currently an employee of Sarepta Therapeutics. J.R.M. is a co-inventor of AAVrh74.MHCK7.micro-dys technology. F.M. has received honoraria and grants from Sarepta Therapeutics for participating at symposia and advisory boards and is involved as an investigator in Sarepta Therapeutics clinical trials. He reports participation in advisory boards for Novartis, F. Hoffmann-La Roche, Ltd., Edgewise Therapeutics, Dyne Therapeutics, Pfizer, PTC Therapeutics and Italfarmaco. C.M.M. reports grants from Capricor Therapeutics, Catabasis, Edgewise Therapeutics, Epirium Bio, Italfarmaco, Pfizer, PTC Therapeutics, Santhera Pharmaceuticals and Sarepta Therapeutics and has a consultancy/advisory role with Biomarin, Capricor Therapeutics, Catalyst, Edgewise Therapeutics, Italfarmaco, PTC Therapeutics, F. Hoffmann-La Roche, Ltd., Santhera Pharmaceuticals and Sarepta Therapeutics. He has received honoraria from PTC Therapeutics and Sarepta Therapeutics. E.M.M. has received fees from AveXis, Biogen and F. Hoffmann-La Roche, Ltd. E.C. has received honoraria from Sarepta Therapeutics for participating in advisory boards and research and/or grant support from the Centers for Disease Control and Prevention, CureSMA, the Muscular Dystrophy Association, the National Institutes of Health, Orphazyme, the Patient-Centered Outcomes Research Institute, Parent Project Muscular Dystrophy, PTC Therapeutics, Santhera, Sarepta Therapeutics and the US Food and Drug Administration. H.K. has received grants from Sarepta Therapeutics, Pfizer, PTC Therapeutics, Taiho Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Nippon Shinyaku Co., Ltd. and Kaneka Corporation. H.K. has received fees from Sarepta Therapeutics, Pfizer, PTC Therapeutics, Chugai Pharmaceutical Co., Nippon Shinyaku Co. and Kaneka Corporation. C.L.-A. is an investigator in Sarepta Therapeutics clinical trials and a sub-investigator in studies sponsored by Pfizer, SolidBioSciences, Edgewise Therapeutics, Italfarmaco and Genentech/Roche. A.N. has received fees from AveXis, Biogen and F. Hoffmann-La Roche, Ltd. C.P. participates on an advisory board and is a consultant for Biogen, Sarepta Therapeutics, AveXis/Novartis Gene Therapies, Genentech/Roche and Scholar Rock; serves as a speaker for Biogen; and is a principal investigator of studies sponsored by AveXis/Novartis Gene Therapies, AMO Pharma, Astellas, Biogen, CSL Behring, Fibrogen, PTC Therapeutics, Pfizer, Sarepta Therapeutics and Scholar Rock. U.S.-S. has received honoraria for counseling and participating in invited talks from Sarepta Therapeutics and F. Hoffmann-La Roche, Ltd. A.V. has a consultancy/advisory role with AMO Pharma, AveXis, Biogen, Edgewise Therapeutics, FibroGen, Novartis, Pfizer, PTC Therapeutics, Sarepta Therapeutics, UCB Pharma, Catalyst and Scholar Rock; has received research funding from AMO Pharma, Capricor Therapeutics, Edgewise Therapeutics, FibroGen, the Muscular Dystrophy Association, Novartis, Parent Project Muscular Dystrophy, Pfizer, RegenxBio and Sarepta Therapeutics; and has other relationship(s) with MedLink Neurology for editorial services. C.M.Z. has received research support from Biogen and Novartis and has served on an advisory board for Sarepta Therapeutics. M.G., C.W. and P.F. are employees of F. Hoffmann-La Roche, Ltd. and may have stock options. A.P.M. and C.R. are employees of Roche Products, Ltd. and may have stock options in F. Hoffmann-La Roche, Ltd. D.R.A., E.D., S.M., R.A.P., T.S., W.Z. and J.S.E. are employees of Sarepta Therapeutics and may have stock options. L.R.R.-K. is an employee of Sarepta Therapeutics and may have stock options. In addition, she is a co-inventor of AAVrh74.MHCK7.micro-dys technology.
Figures


References
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
