

This is a preprint.
Location-biased β-arrestin conformations direct GPCR signaling
- PMID: 39386521
- PMCID: PMC11463559
- DOI: 10.1101/2024.09.24.614742
Location-biased β-arrestin conformations direct GPCR signaling
Abstract
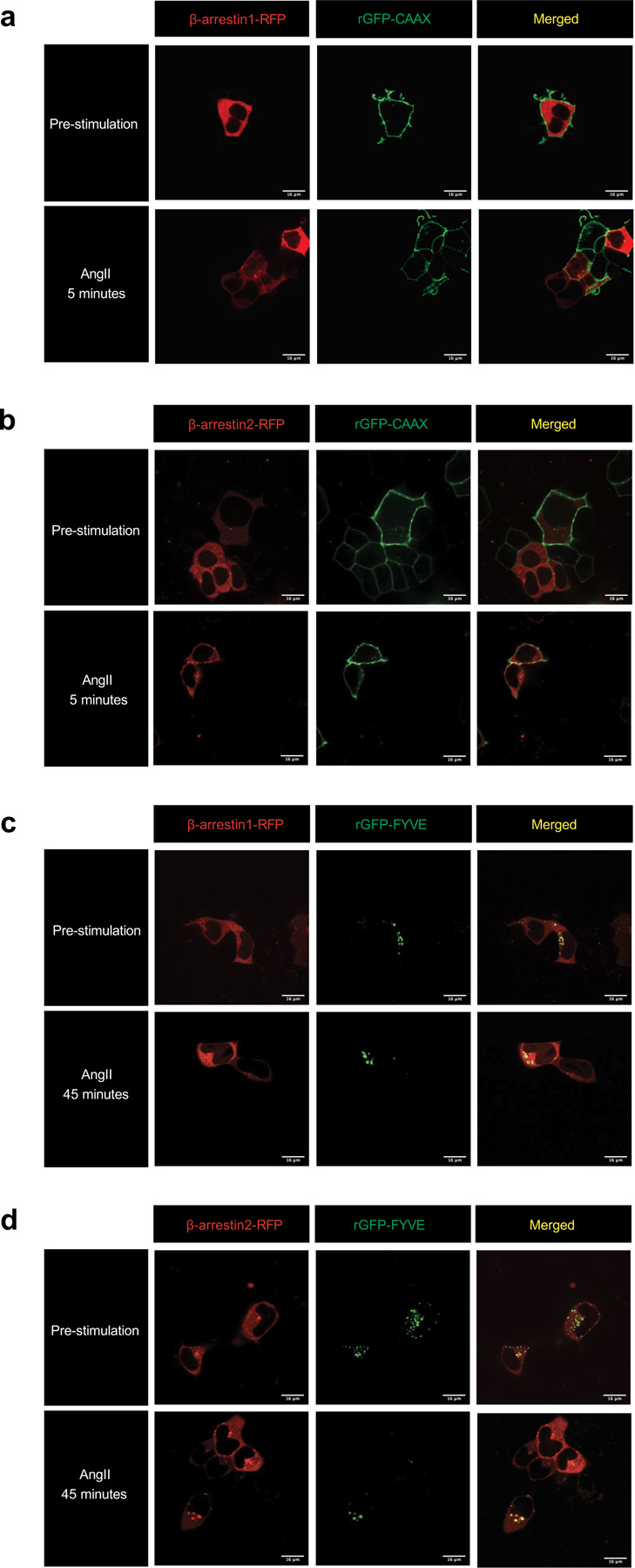
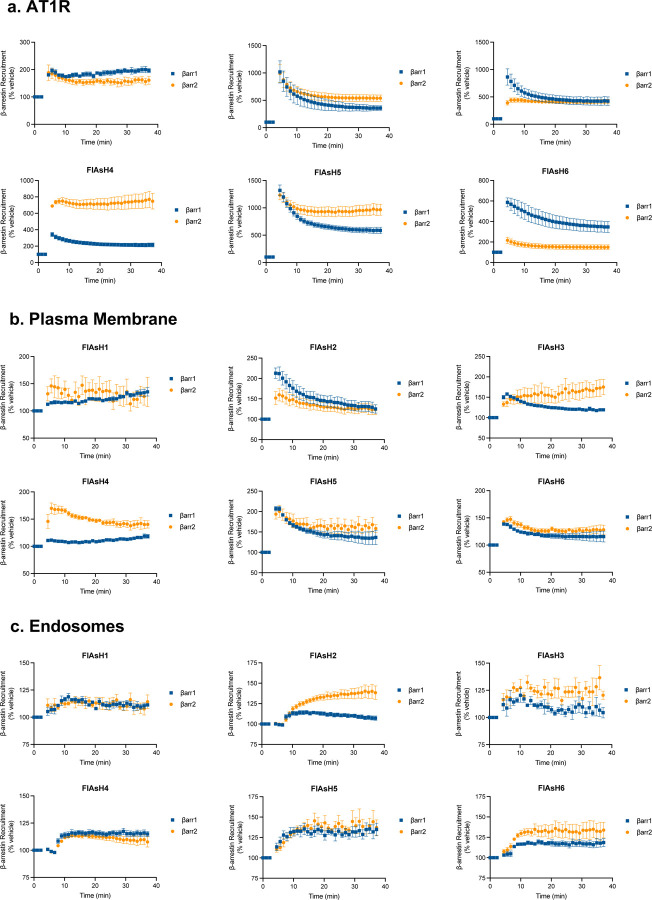
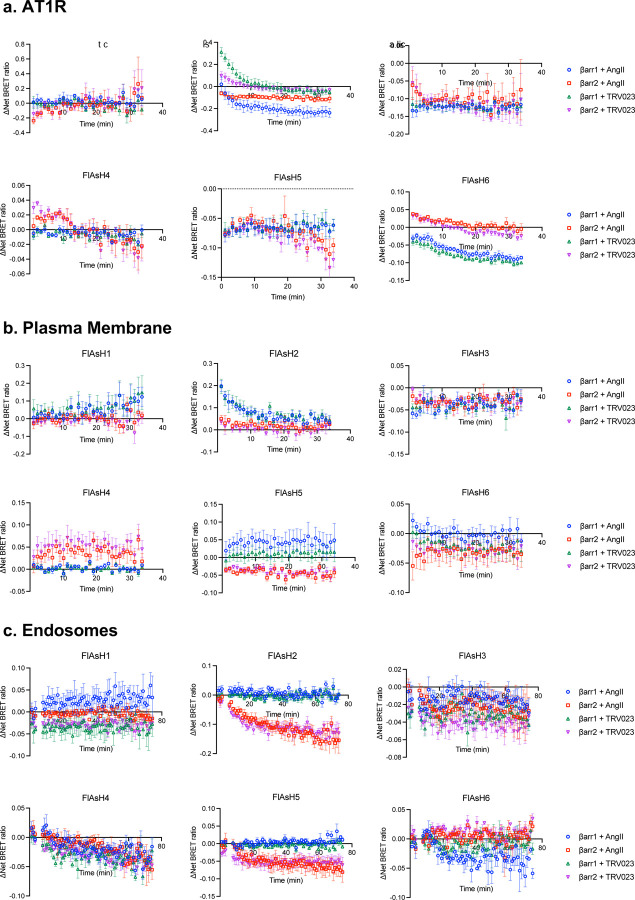
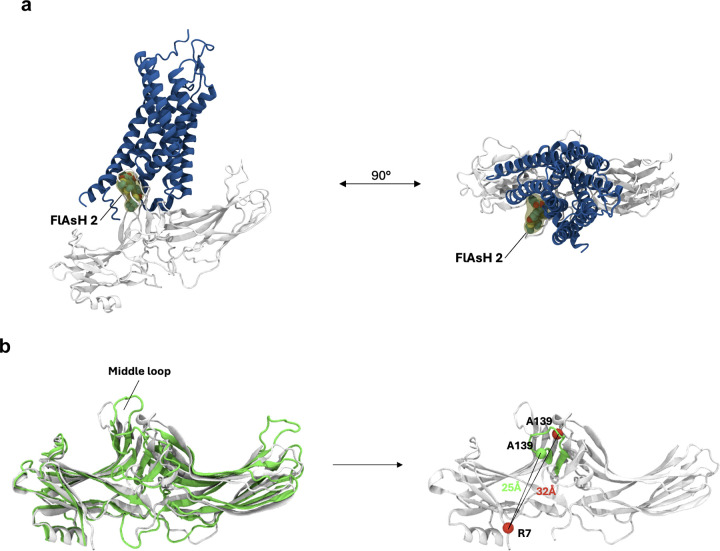
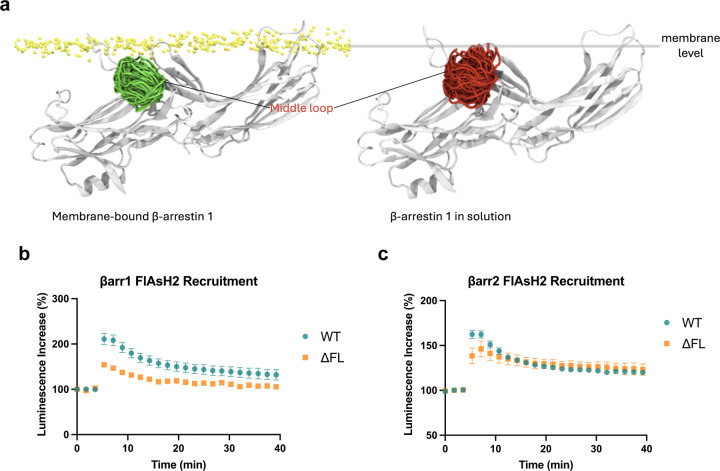
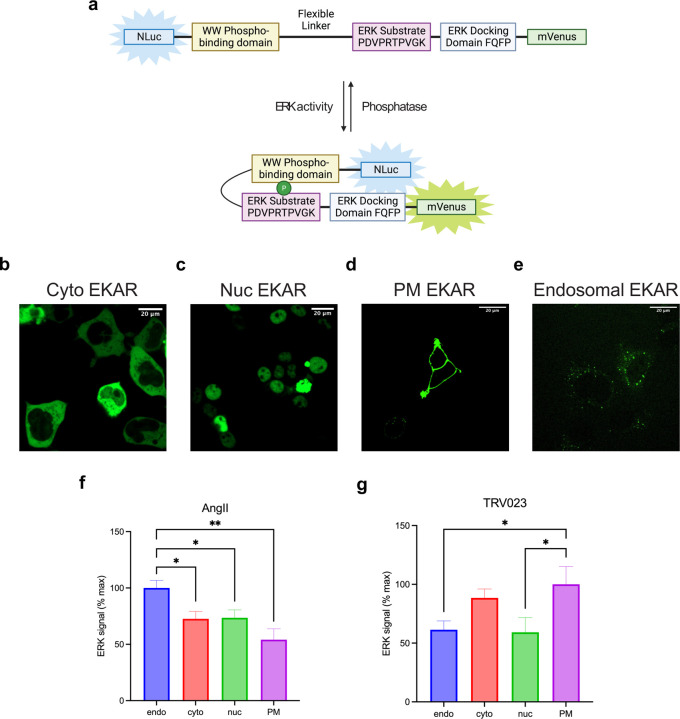
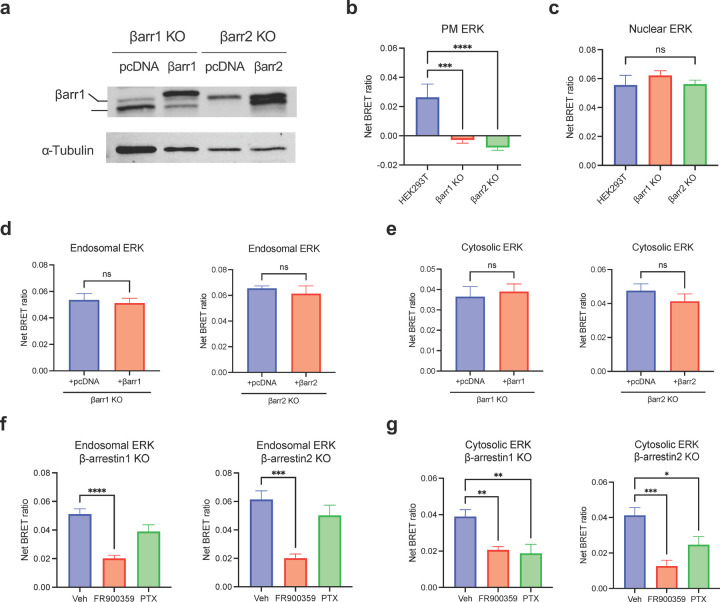
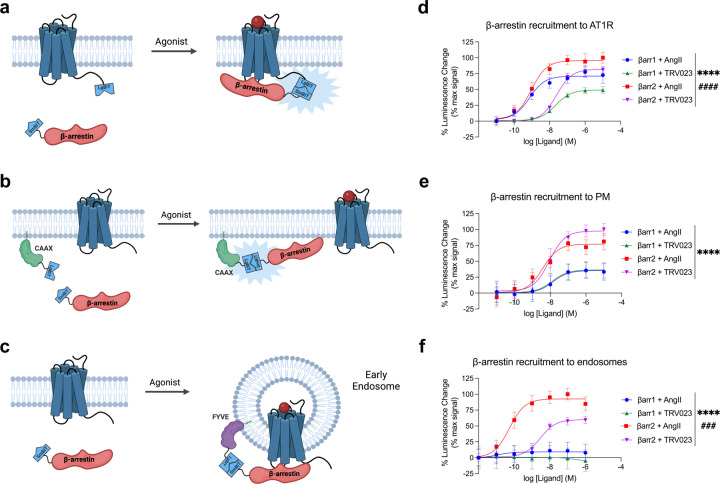
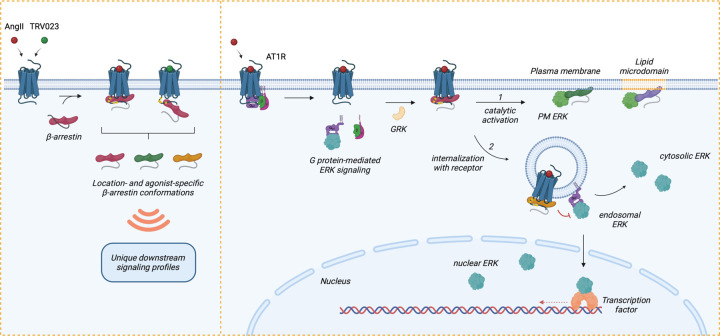
β-arrestins are multifunctional intracellular proteins that regulate the desensitization, internalization and signaling of over 800 different G protein-coupled receptors (GPCRs) and interact with a diverse array of cellular partners1,2. Beyond the plasma membrane, GPCRs can initiate unique signaling cascades from various subcellular locations, a phenomenon known as "location bias"3,4. Here, we investigate how β-arrestins direct location-biased signaling of the angiotensin II type I receptor (AT1R). Using novel bioluminescence resonance energy transfer (BRET) conformational biosensors and extracellular signal-regulated kinase (ERK) activity reporters, we reveal that in response to the endogenous agonist Angiotensin II and the β-arrestin-biased agonist TRV023, β-arrestin 1 and β-arrestin 2 adopt distinct conformations across different subcellular locations, which are intricately linked to differential ERK activation profiles. We also uncover a population of receptor-free catalytically activated β-arrestins in the plasma membrane that exhibits insensitivity to different agonists and promotes ERK activation on the plasma membrane independent of G proteins. These findings deepen our understanding of GPCR signaling complexity and also highlight the nuanced roles of β-arrestins beyond traditional G protein pathways.
Keywords: FlAsH; G protein-coupled receptor; GPCR; MAP kinase; beta-arrestin; biased agonism; biased signaling; biosensors; catalytic activation; conformations; location bias.
Conflict of interest statement
DECLARATION OF INTERESTS The authors declare no competing interests.
Figures
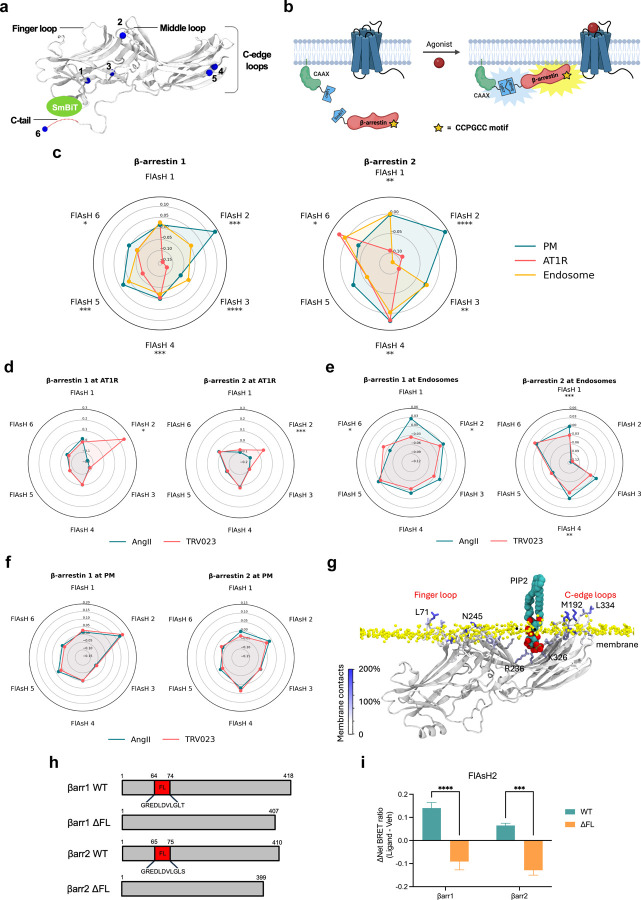
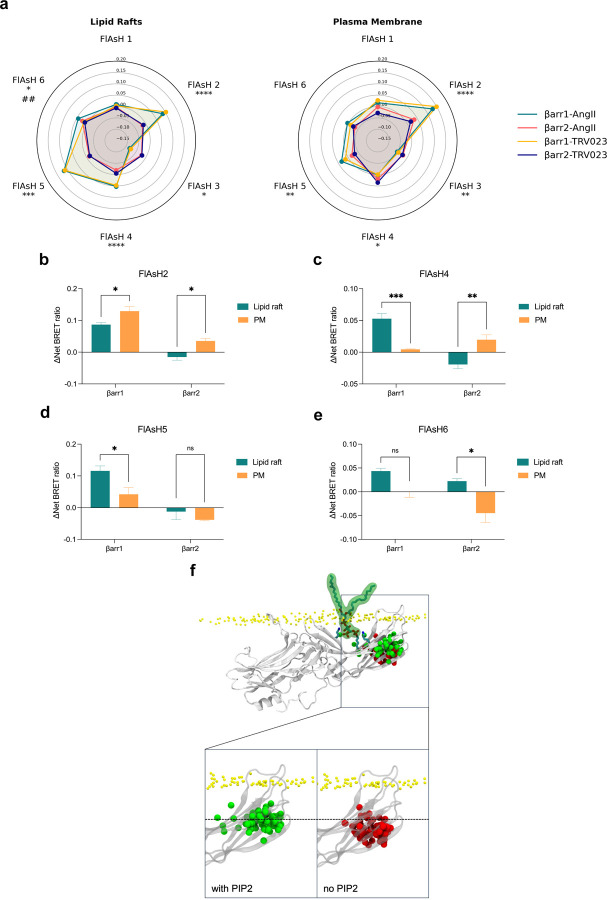
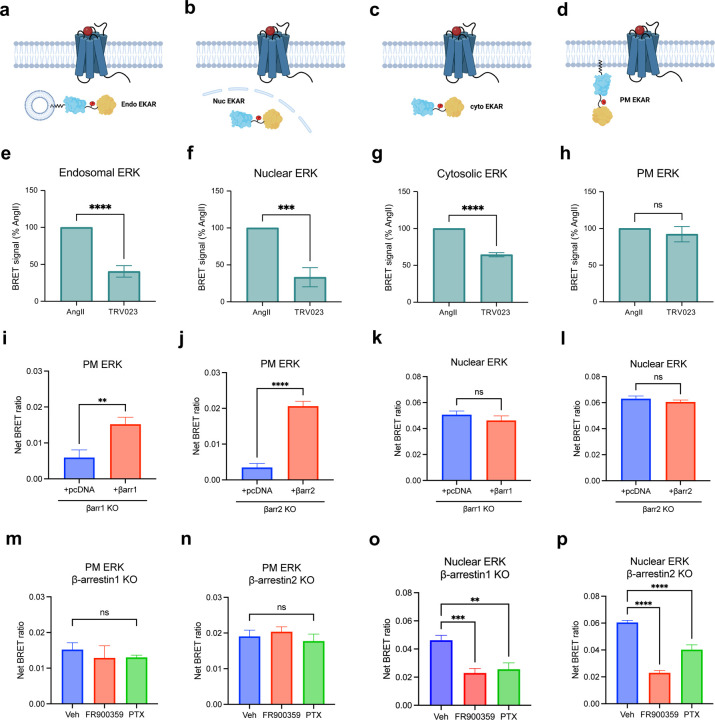
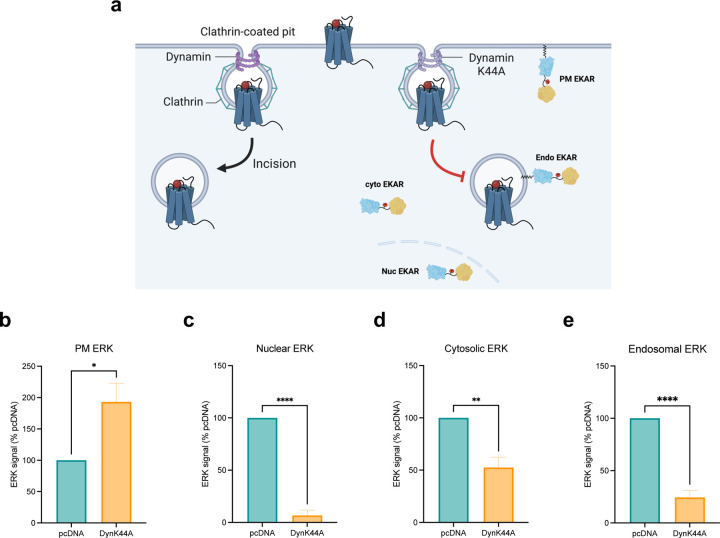
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous