

The conserved genetic program of male germ cells uncovers ancient regulators of human spermatogenesis
- PMID: 39388236
- PMCID: PMC11466473
- DOI: 10.7554/eLife.95774
The conserved genetic program of male germ cells uncovers ancient regulators of human spermatogenesis
Abstract
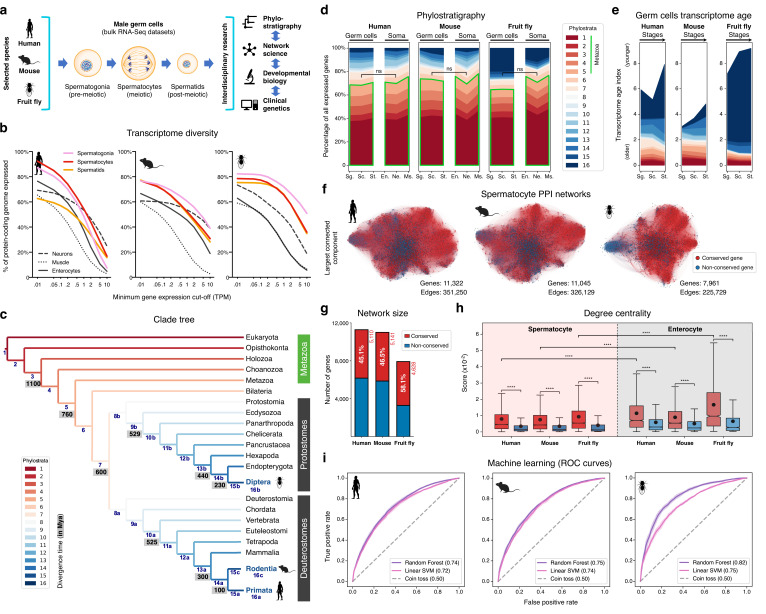
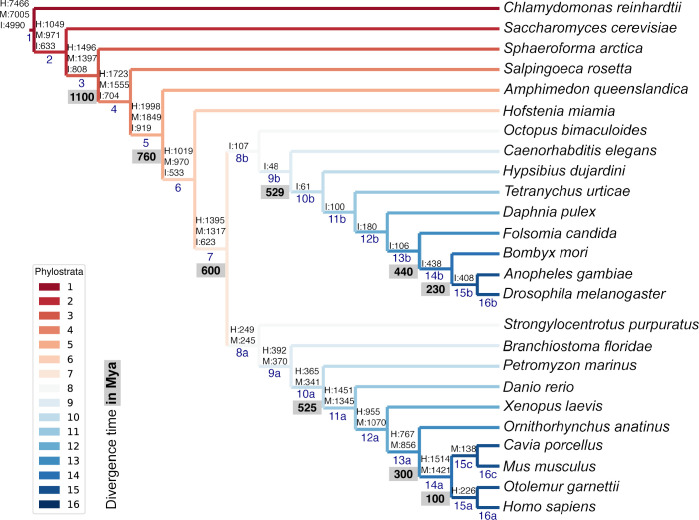
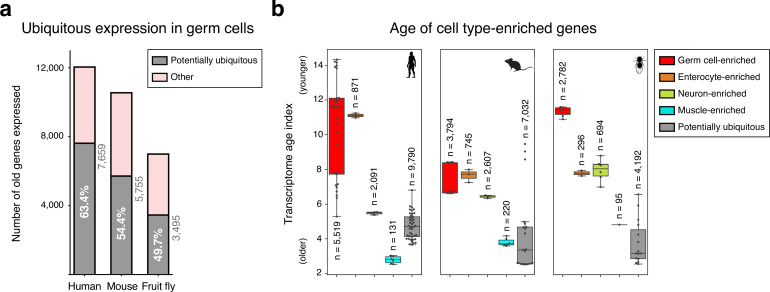
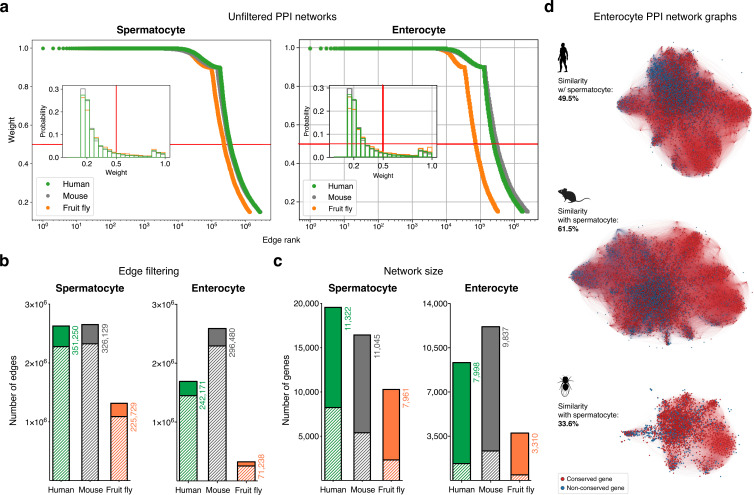
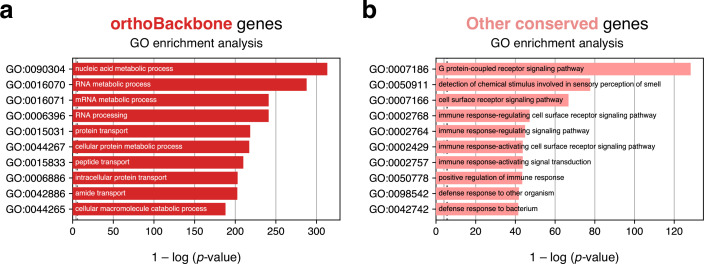
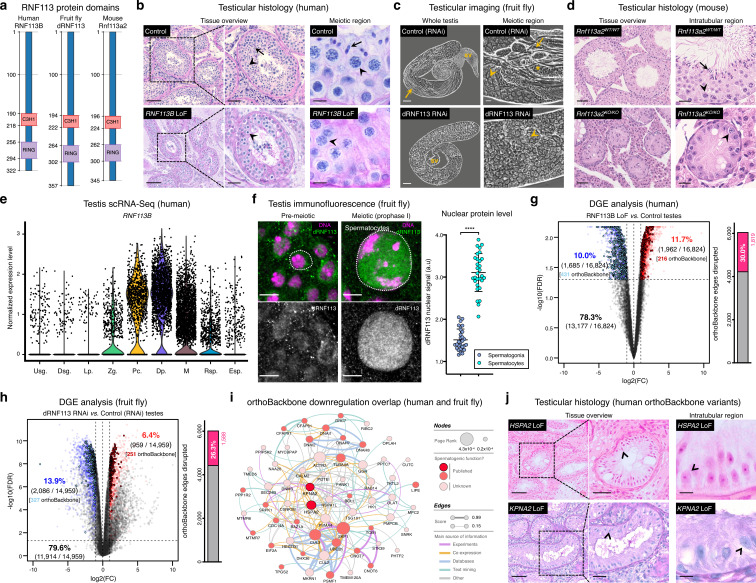
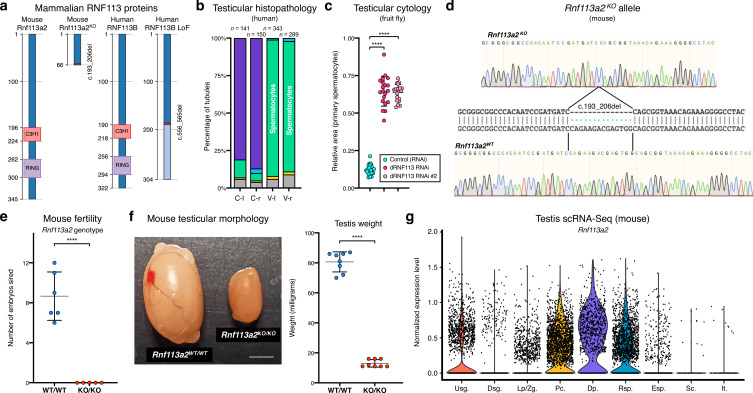
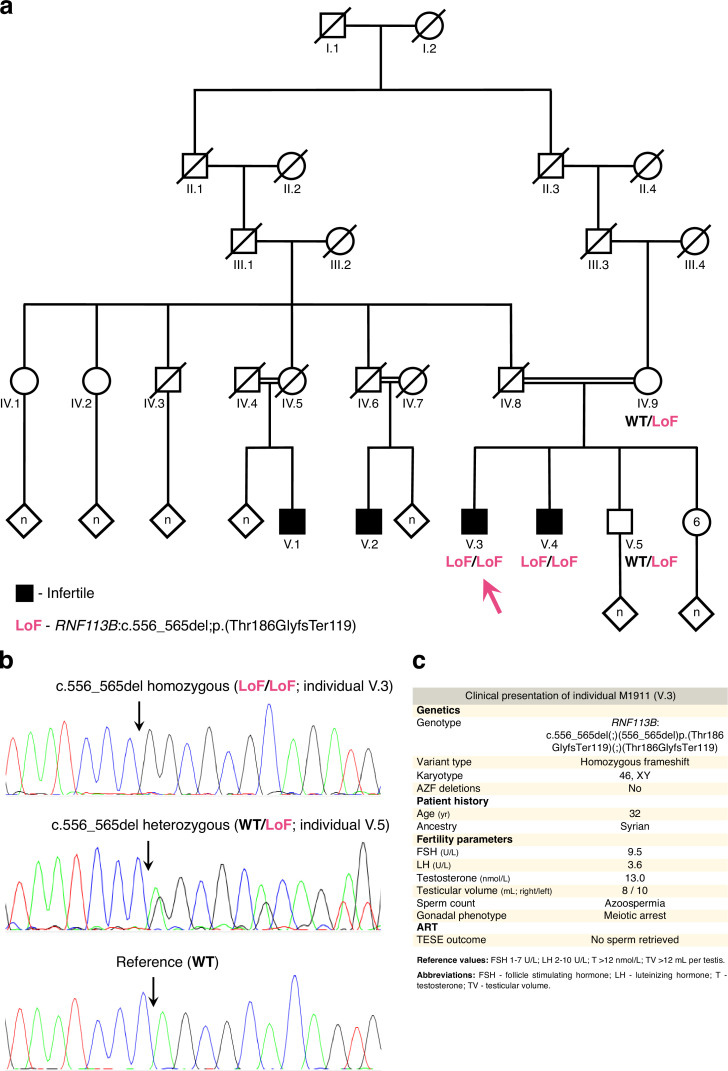
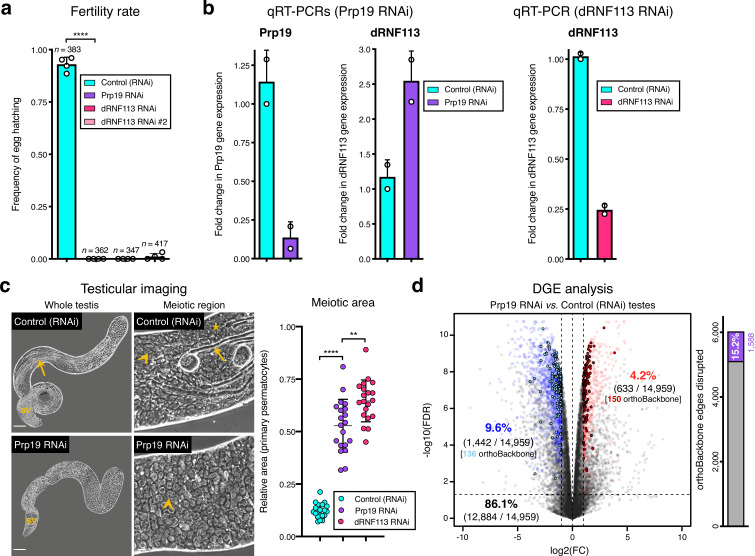
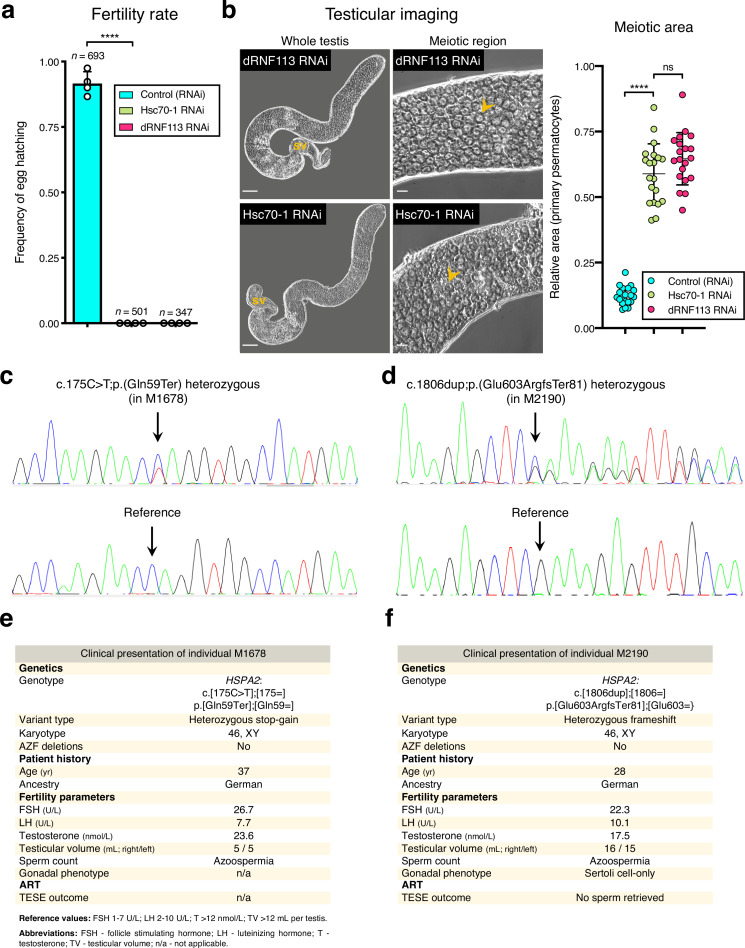
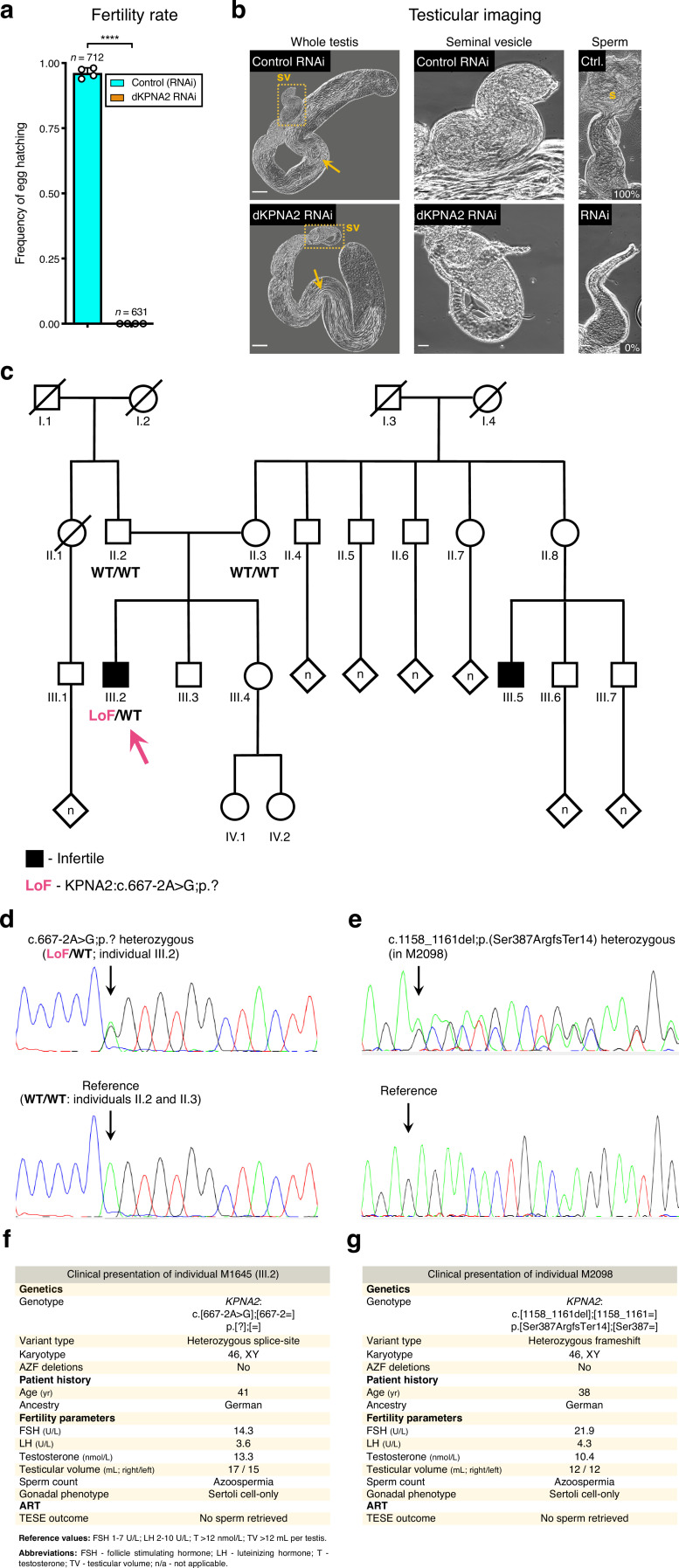
Male germ cells share a common origin across animal species, therefore they likely retain a conserved genetic program that defines their cellular identity. However, the unique evolutionary dynamics of male germ cells coupled with their widespread leaky transcription pose significant obstacles to the identification of the core spermatogenic program. Through network analysis of the spermatocyte transcriptome of vertebrate and invertebrate species, we describe the conserved evolutionary origin of metazoan male germ cells at the molecular level. We estimate the average functional requirement of a metazoan male germ cell to correspond to the expression of approximately 10,000 protein-coding genes, a third of which defines a genetic scaffold of deeply conserved genes that has been retained throughout evolution. Such scaffold contains a set of 79 functional associations between 104 gene expression regulators that represent a core component of the conserved genetic program of metazoan spermatogenesis. By genetically interfering with the acquisition and maintenance of male germ cell identity, we uncover 161 previously unknown spermatogenesis genes and three new potential genetic causes of human infertility. These findings emphasize the importance of evolutionary history on human reproductive disease and establish a cross-species analytical pipeline that can be repurposed to other cell types and pathologies.
Keywords: D. melanogaster; evolutionary biology; gene expression; germ cells; human; infertility; medicine; meiosis; mouse; spermatogenesis.
Plain language summary
Sperm are one of the most remarkable cells in nature, safely housing genetic information while also often moving through foreign environments in search of an egg to fertilize. Central for sexual reproduction, sperm cells of all shapes and sizes are found in animals, plants and even some species of fungi. You may be familiar with the streamlined structure of human sperm, for example, with its round head and flexible tail; but the sperm cells of fruit flies are about 300 times longer, and those found in mice have a hook-shaped head. Relatedly, the genes involved in the creation of reproductive cells often show rapid evolution, with their sequences quickly diverging between species. Due to the complexity of the network of genetic interactions taking place during sperm development, it has so far been difficult to fully isolate the ‘core program’ that governs sperm assembly and allows these cells to acquire their distinct identity. Whether this program could be conserved and shared across the tree of life, in particular, remains unclear. In response, Brattig-Correia, Almeida, Wyrwoll et al. first conducted analyses that allowed them to pinpoint the genes that were ‘switched on’ during the formation of human, mouse and fruit fly sperm. Assessing the ‘age’ of these genes showed that a large proportion had emerged early during evolution. Shared across the three species, these deeply conserved genes were shown to play a fundamental role in sperm cells acquiring and maintaining their identity. Further genetic experiments were conducted in fruit flies to refine these findings, highlighting a set of 161 previously unknown genes essential for sperm formation. By combining these results with genetic data from men unable to have children, Brattig-Correia, Almeida, Wyrwoll et al. were able to identify three new genes that could play a role in human infertility. This work emphasizes how our understanding of human reproductive development can benefit from examining this process in other species, and its evolutionary history. In particular, the knowledge gained from these comparative approaches could ultimately help develop better genetic tests and treatments for human infertility.
© 2024, Brattig-Correia, Almeida, Wyrwoll et al.
Conflict of interest statement
RB, JA, MW, IJ, DS, CM, SD, LG, HS, NS, PP, AN, AL, JB, SL, MM, SK, MM, LR, FT, JB, PN No competing interests declared
Figures
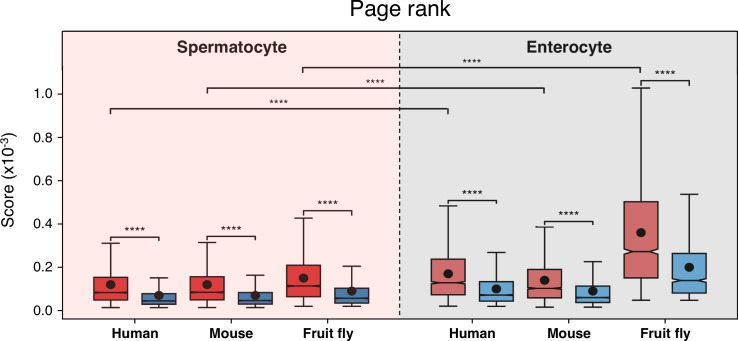
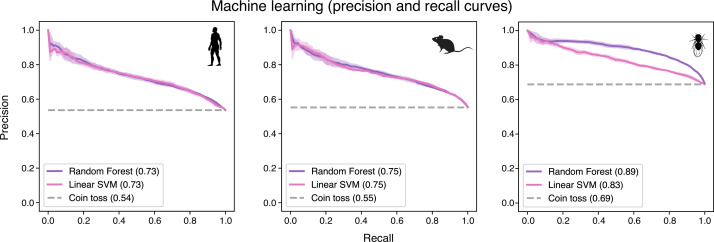
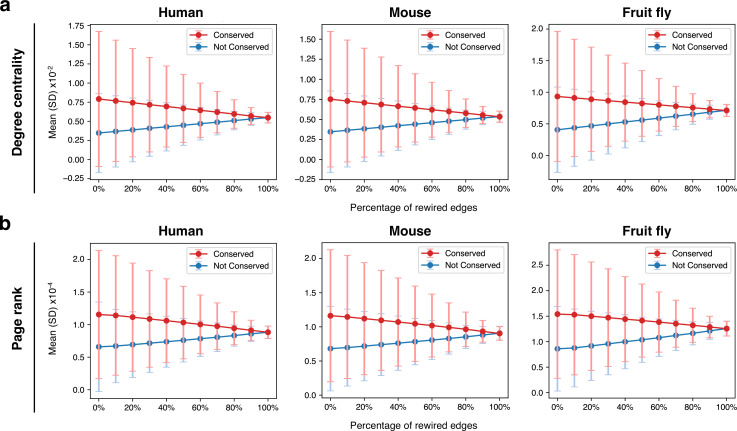
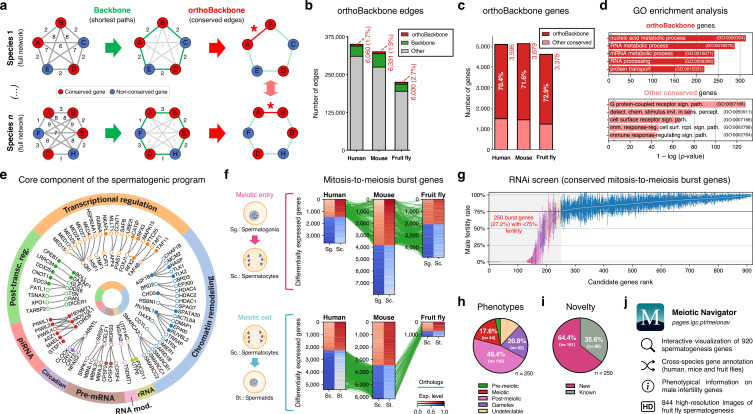

Update of
- doi: 10.1101/2022.03.02.482557
- doi: 10.7554/eLife.95774.1
- doi: 10.7554/eLife.95774.2
References
-
- Baldarelli RM, Smith CM, Finger JH, Hayamizu TF, McCright IJ, Xu J, Shaw DR, Beal JS, Blodgett O, Campbell J, Corbani LE, Frost PJ, Giannatto SC, Miers DB, Kadin JA, Richardson JE, Ringwald M. The mouse gene expression database (GXD): 2021 update. Nucleic Acids Research. 2021;49:D924–D931. doi: 10.1093/nar/gkaa914. - DOI - PMC - PubMed
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
