

NAD+ depletion is central to placental dysfunction in an inflammatory subclass of preeclampsia
- PMID: 39389781
- PMCID: PMC11467044
- DOI: 10.26508/lsa.202302505
NAD+ depletion is central to placental dysfunction in an inflammatory subclass of preeclampsia
Abstract
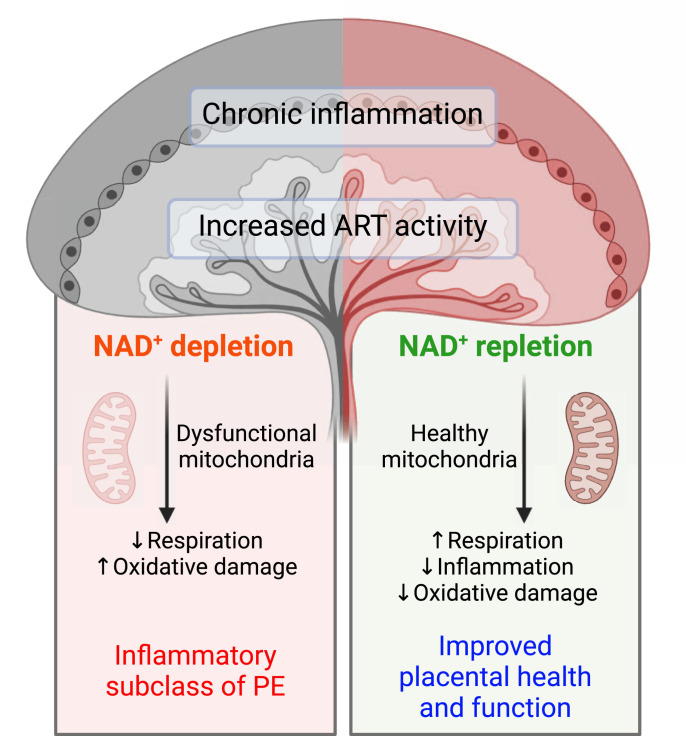
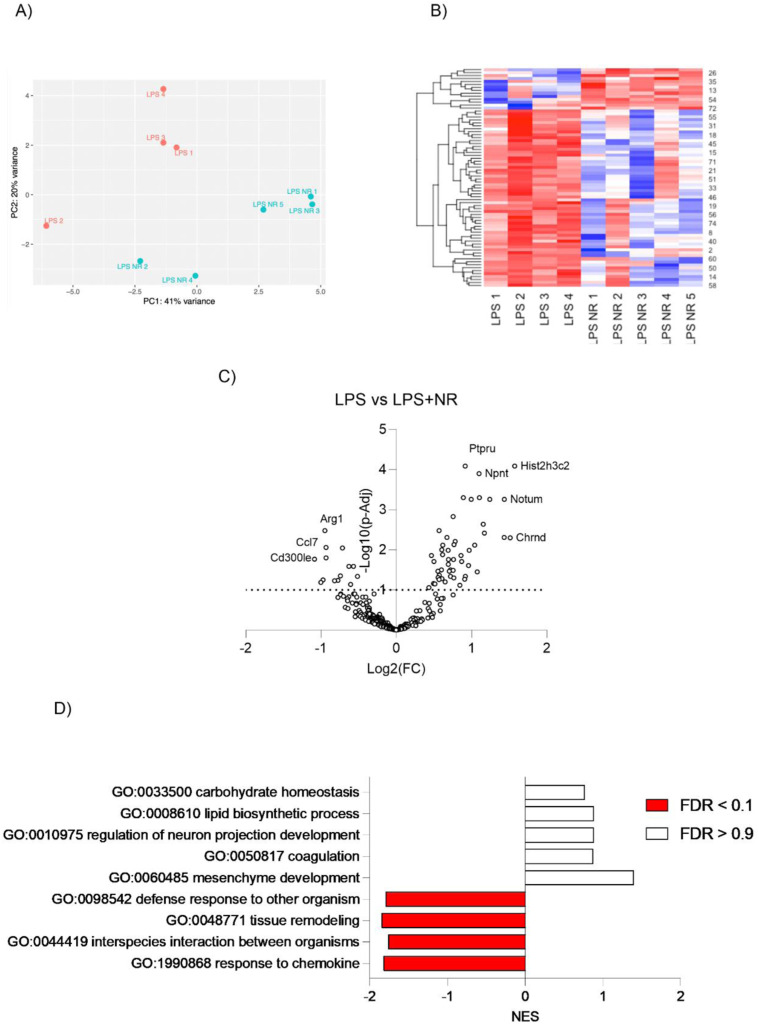
Preeclampsia (PE) is a hypertensive disorder of pregnancy and a major cause of maternal/perinatal adverse health outcomes with no effective therapeutic strategies. Our group previously identified distinct subclasses of PE, one of which exhibits heightened placental inflammation (inflammation-driven PE). In non-pregnant populations, chronic inflammation is associated with decreased levels of cellular NAD+, a vitamin B3 derivative involved in energy metabolism and mitochondrial function. Interestingly, specifically in placentas from women with inflammation-driven PE, we observed the increased activity of NAD+-consuming enzymes, decreased NAD+ content, decreased expression of mitochondrial proteins, and increased oxidative damage. HTR8 human trophoblasts likewise demonstrated increased NAD+-dependent ADP-ribosyltransferase (ART) activity, coupled with decreased mitochondrial respiration rates and invasive function under inflammatory conditions. Such adverse effects were attenuated by boosting cellular NAD+ levels with nicotinamide riboside (NR). Finally, in an LPS-induced rat model of inflammation-driven PE, NR administration (200 mg/kg/day) from gestational days 1-19 prevented maternal hypertension and fetal/placental growth restriction, improved placental mitochondrial function, and reduced inflammation and oxidative stress. This study demonstrates the critical role of NAD+ in maintaining placental function and identifies NAD+ boosting as a promising preventative strategy for PE.
© 2024 Jahan et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures







References
-
- Powers RW, Roberts JM, Plymire DA, Pucci D, Datwyler SA, Laird DM, Sogin DC, Jeyabalan A, Hubel CA, Gandley RE (2012) Low placental growth factor across pregnancy identifies a subset of women with preterm preeclampsia: Type 1 versus type 2 preeclampsia? Hypertension 60: 239–246. 10.1161/HYPERTENSIONAHA.112.191213 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources