

Human DNA polymerase ε is a source of C>T mutations at CpG dinucleotides
- PMID: 39390083
- PMCID: PMC11549043
- DOI: 10.1038/s41588-024-01945-x
Human DNA polymerase ε is a source of C>T mutations at CpG dinucleotides
Abstract
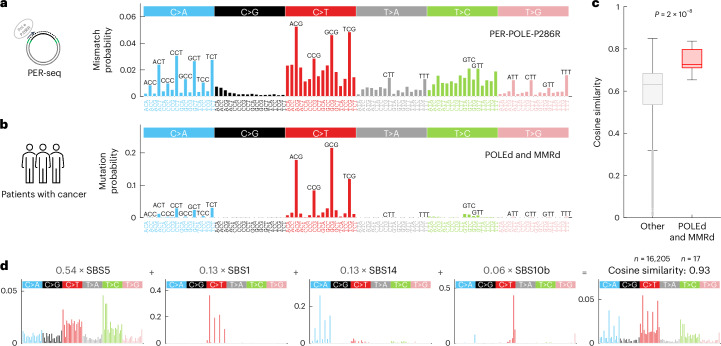
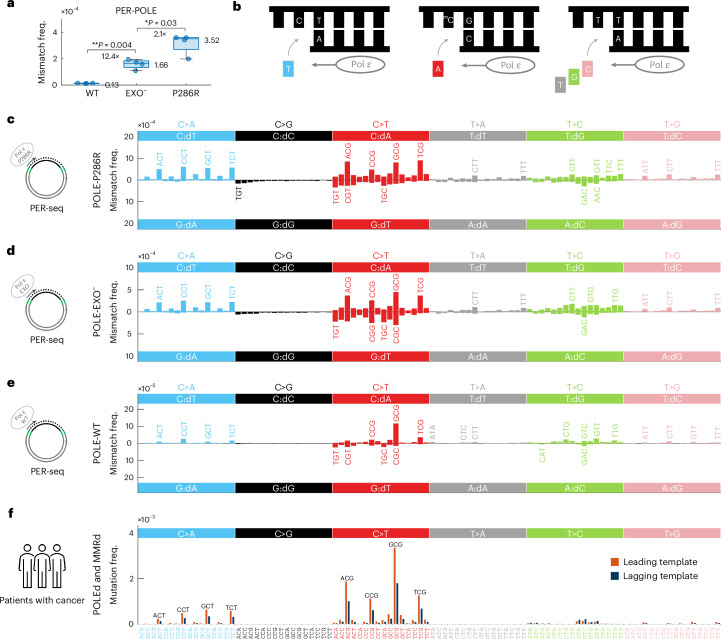
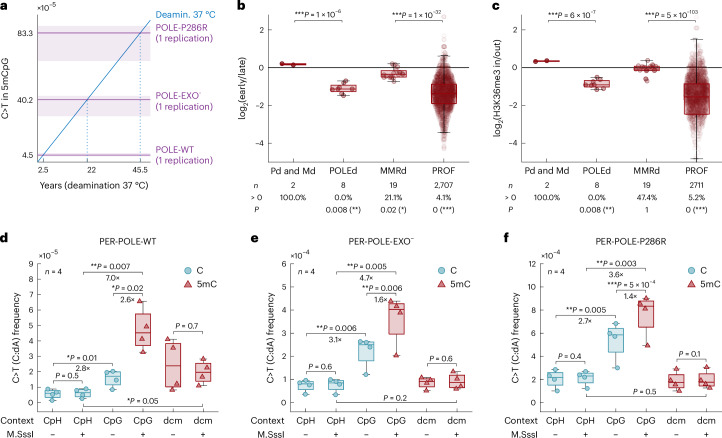
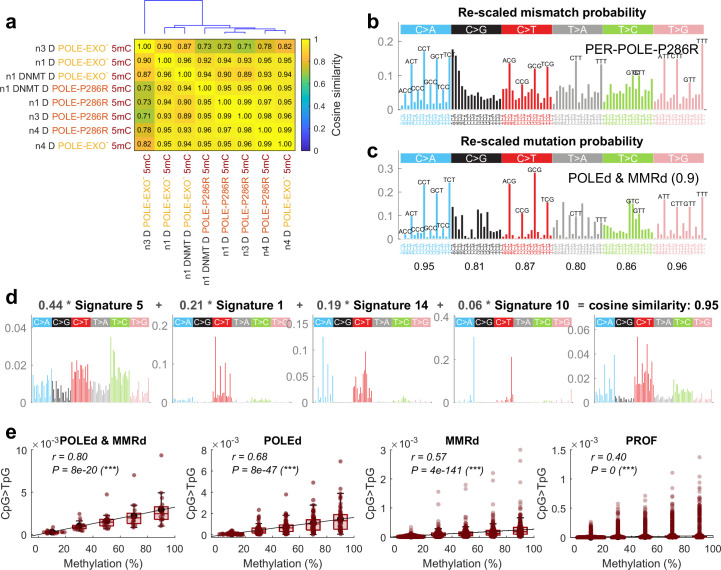
C-to-T transitions in CpG dinucleotides are the most prevalent mutations in human cancers and genetic diseases. These mutations have been attributed to deamination of 5-methylcytosine (5mC), an epigenetic modification found on CpGs. We recently linked CpG>TpG mutations to replication and hypothesized that errors introduced by polymerase ε (Pol ε) may represent an alternative source of mutations. Here we present a new method called polymerase error rate sequencing (PER-seq) to measure the error spectrum of DNA polymerases in isolation. We find that the most common human cancer-associated Pol ε mutant (P286R) produces an excess of CpG>TpG errors, phenocopying the mutation spectrum of tumors carrying this mutation and deficiencies in mismatch repair. Notably, we also discover that wild-type Pol ε has a sevenfold higher error rate when replicating 5mCpG compared to C in other contexts. Together, our results from PER-seq and human cancers demonstrate that replication errors are a major contributor to CpG>TpG mutagenesis in replicating cells, fundamentally changing our understanding of this important disease-causing mutational mechanism.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures









References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
