

Enterococcal-host interactions in the gastrointestinal tract and beyond
- PMID: 39391373
- PMCID: PMC11466040
- DOI: 10.1093/femsmc/xtae027
Enterococcal-host interactions in the gastrointestinal tract and beyond
Abstract
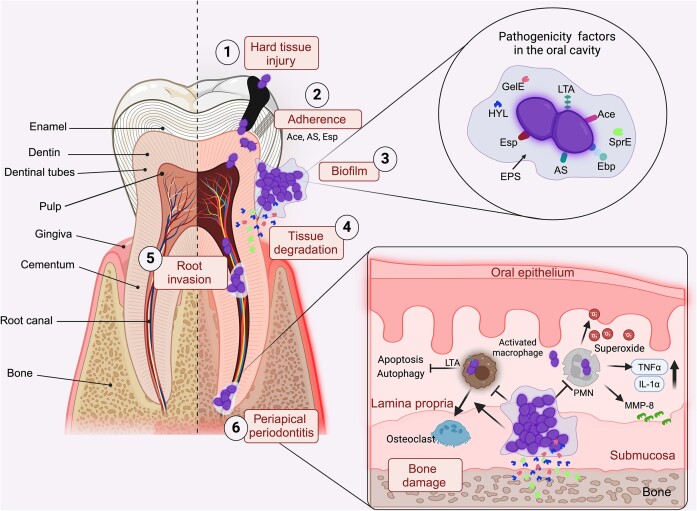
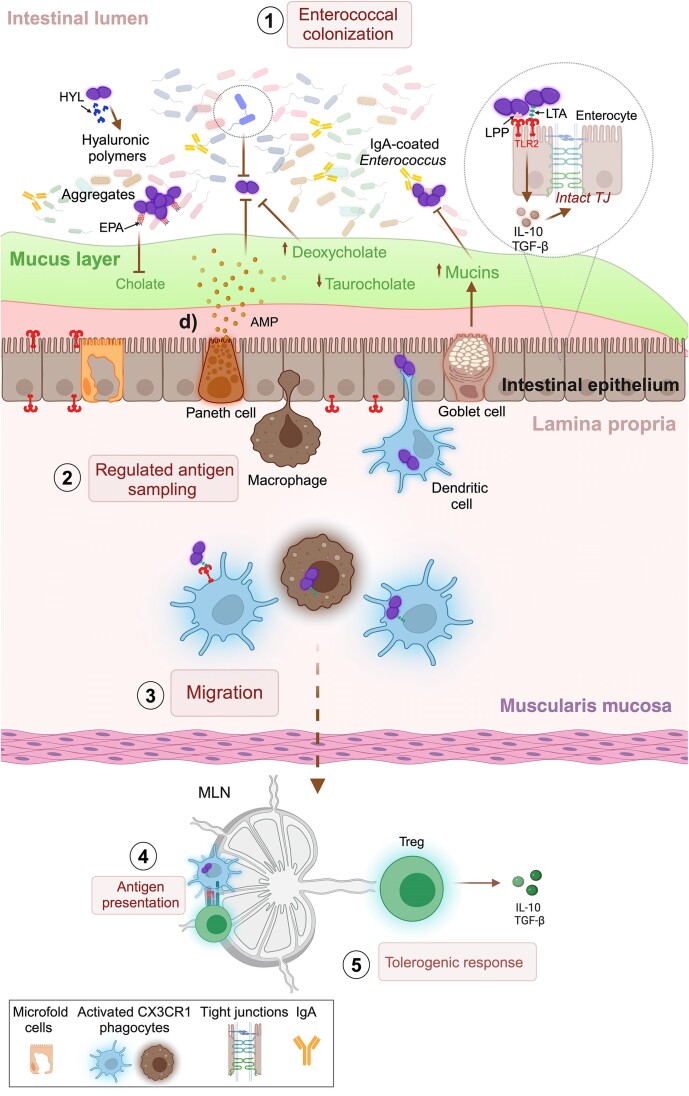
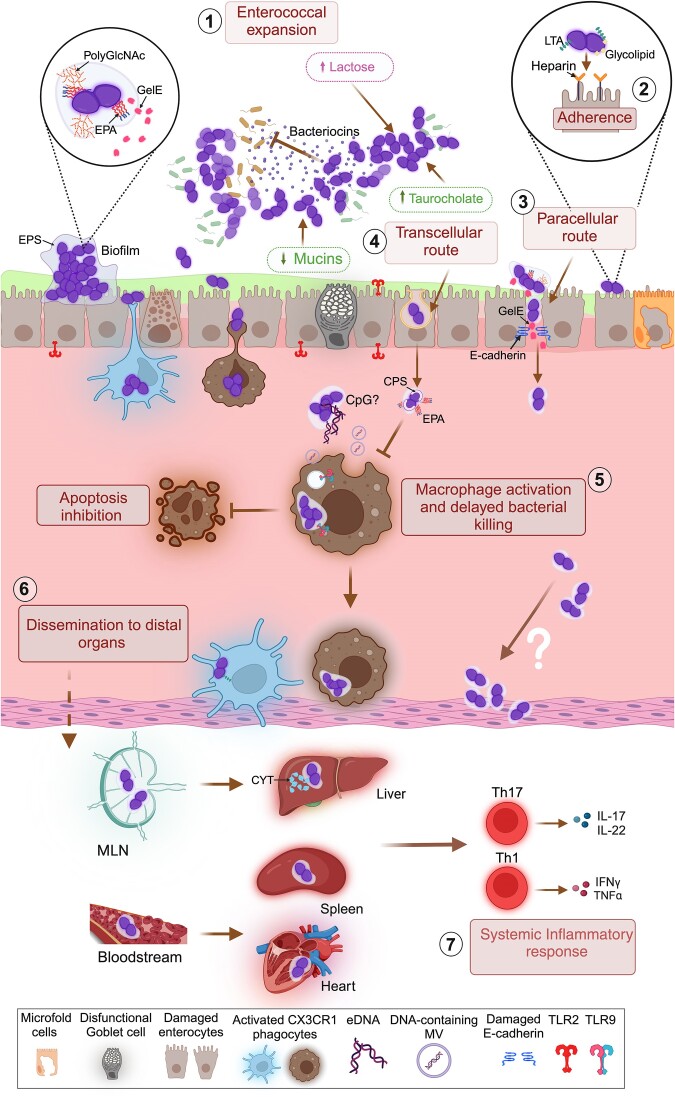
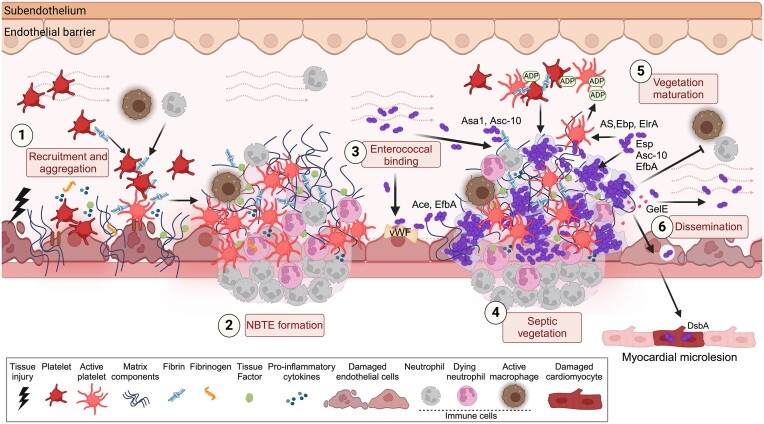
The gastrointestinal tract (GIT) is typically considered the natural niche of enterococci. However, these bacteria also inhabit extraintestinal tissues, where they can disrupt organ physiology and cause life-threatening infections. Here, we discuss how enterococci, primarily Enterococcus faecalis, interact with the intestine and other host anatomical locations such as the oral cavity, heart, liver, kidney, and vaginal tract. The metabolic flexibility of these bacteria allows them to quickly adapt to new environments, promoting their persistence in diverse tissues. In transitioning from commensals to pathogens, enterococci must overcome harsh conditions such as nutrient competition, exposure to antimicrobials, and immune pressure. Therefore, enterococci have evolved multiple mechanisms to adhere, colonize, persist, and endure these challenges in the host. This review provides a comprehensive overview of how enterococci interact with diverse host cells and tissues across multiple organ systems, highlighting the key molecular pathways that mediate enterococcal adaptation, persistence, and pathogenic behavior.
Keywords: commensal to pathogen transition; dysbiosis; enterococcal-host interactions; enterococci; gastrointestinal tract; inter-organ dissemination.
© The Author(s) 2024. Published by Oxford University Press on behalf of FEMS.
Conflict of interest statement
None declared.
Figures
References
-
- Agudelo Higuita NI, Huycke MM. Enterococcal disease, epidemiology, and implications for treatment. In: Gilmore MS, Clewell DB, Ike Y, Shankar N (eds), Enterococci: From Commensals to Leading Causes of Drug Resistant Infection. Boston: Massachusetts Eye and Ear Infirmary, 2014.
-
- Akbari Aghdam M, Soroush Barhaghi MH, Aghazadeh M et al. Virulence genes in biofilm producer Enterococcus faecalis isolates from root canal infections. Cell Mol Biol (Noisy-le-grand). 2017;63:55–59. - PubMed
-
- Al-Ahmad A, Muller N, Wiedmann-Al-Ahmad M et al. Endodontic and salivary isolates of Enterococcus faecalis integrate into biofilm from human salivary bacteria cultivated in vitro. J Endod. 2009;35:986–91. - PubMed
Publication types
LinkOut - more resources
Full Text Sources