

Dysregulation of protein SUMOylation networks in Huntington's disease R6/2 mouse striatum
- PMID: 39391934
- PMCID: PMC11969464
- DOI: 10.1093/brain/awae319
Dysregulation of protein SUMOylation networks in Huntington's disease R6/2 mouse striatum
Abstract
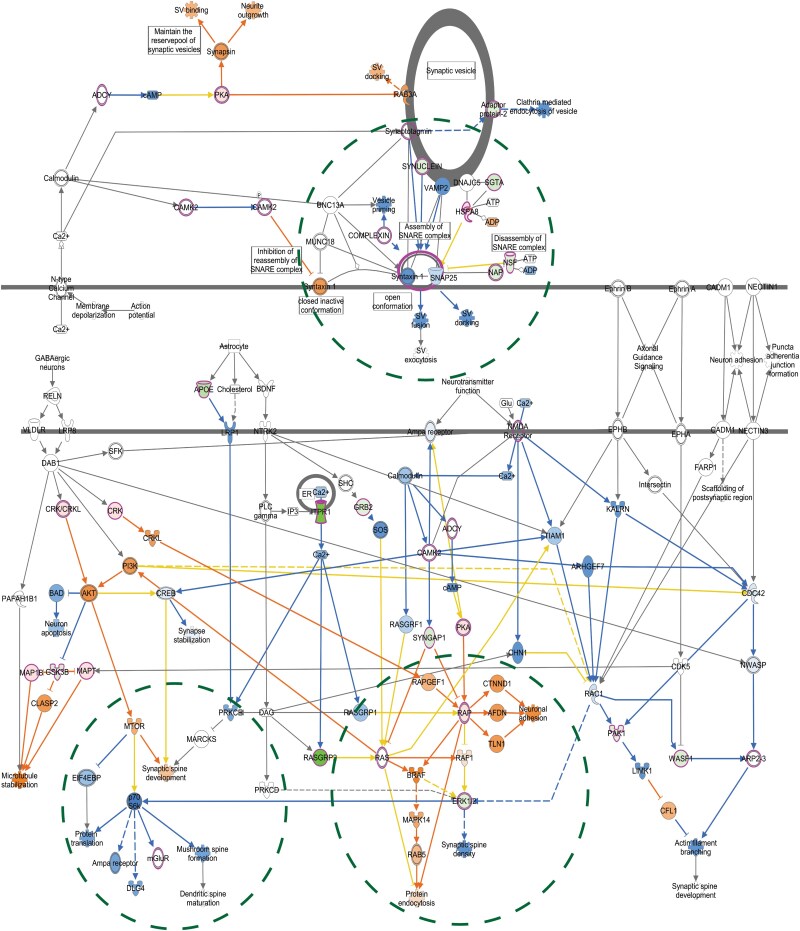
Huntington's disease is a neurodegenerative disorder caused by an expanded CAG repeat mutation in the Huntingtin (HTT) gene. The mutation impacts neuronal protein homeostasis and cortical/striatal circuitry. SUMOylation is a post-translational modification with broad cellular effects including via modification of synaptic proteins. Here, we used an optimized SUMO protein-enrichment and mass spectrometry method to identify the protein SUMOylation/SUMO interaction proteome in the context of Huntington's disease using R6/2 transgenic and non-transgenic mice. Significant changes in the enrichment of SUMOylated and SUMO-interacting proteins were observed, including those involved in presynaptic function, cytomatrix at the active zone, cytoskeleton organization and glutamatergic signalling. Mitochondrial and RNA-binding proteins also showed altered enrichment. Modified SUMO-associated pathways in Huntington's disease tissue include clathrin-mediated endocytosis signalling, synaptogenesis signalling, synaptic long-term potentiation and SNARE signalling. To evaluate how modulation of SUMOylation might influence functional measures of neuronal activity in Huntington's disease cells in vitro, we used primary neuronal cultures from R6/2 and non-transgenic mice. A receptor internalization assay for the metabotropic glutamate receptor 7 (mGLUR7), a SUMO-enriched protein in the mass spectrometry, showed decreased internalization in R6/2 neurons compared to non-transgenic neurons. SiRNA-mediated knockdown of the E3 SUMO ligase protein inhibitor of activated STAT1 (Pias1), which can SUMO modify mGLUR7, reduced this Huntington's disease phenotype. In addition, microelectrode array analysis of primary neuronal cultures indicated early hyperactivity in Huntington's disease cells, while later time points demonstrated deficits in several measurements of neuronal activity within cortical neurons. Huntington's disease phenotypes were rescued at selected time points following knockdown of Pias1. Collectively, our results provide a mouse brain SUMOome resource and show that significant alterations occur within the post-translational landscape of SUMO-protein interactions of synaptic proteins in Huntington's disease mice, suggesting that targeting of synaptic SUMO networks may provide a proteostatic systems-based therapeutic approach for Huntington's disease and other neurological disorders.
Keywords: E3 SUMO ligase; Huntington’s disease; PIAS1; SUMO; mGLUR7; proteomics; synaptic.
© The Author(s) 2024. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
The authors report no competing interests.
Figures





References
-
- Huntington’s Disease Collaborative Research Group . A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. - PubMed
-
- Li H, Wyman T, Yu ZX, Li SH, Li XJ. Abnormal association of mutant huntingtin with synaptic vesicles inhibits glutamate release. Hum Mol Genet. 2003;12:2021–2030. - PubMed
-
- Vitet H, Brandt V, Saudou F. Traffic signaling: New functions of huntingtin and axonal transport in neurological disease. Curr Opin Neurobiol. 2020;63:122–130. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
