

Modeling of mRNA deadenylation rates reveal a complex relationship between mRNA deadenylation and decay
- PMID: 39394354
- PMCID: PMC11649921
- DOI: 10.1038/s44318-024-00258-3
Modeling of mRNA deadenylation rates reveal a complex relationship between mRNA deadenylation and decay
Abstract
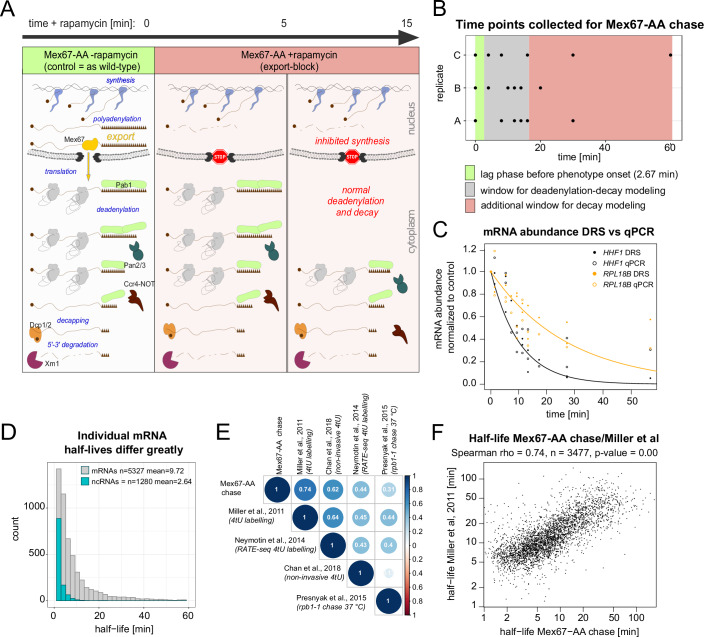
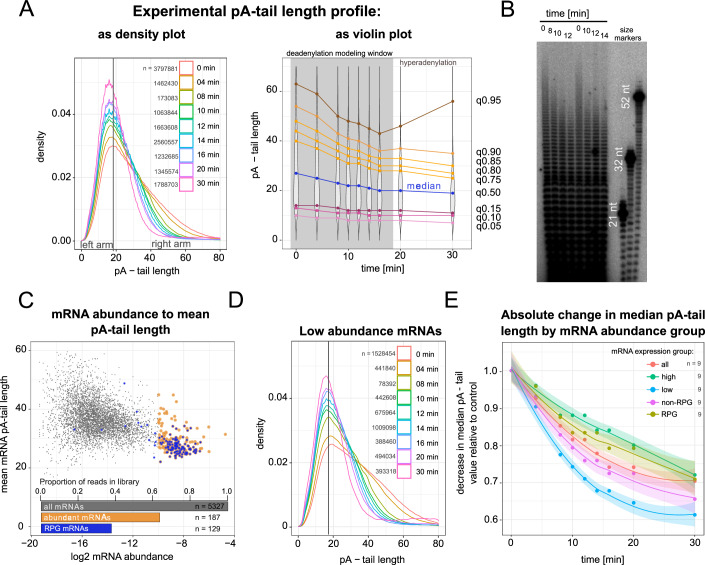
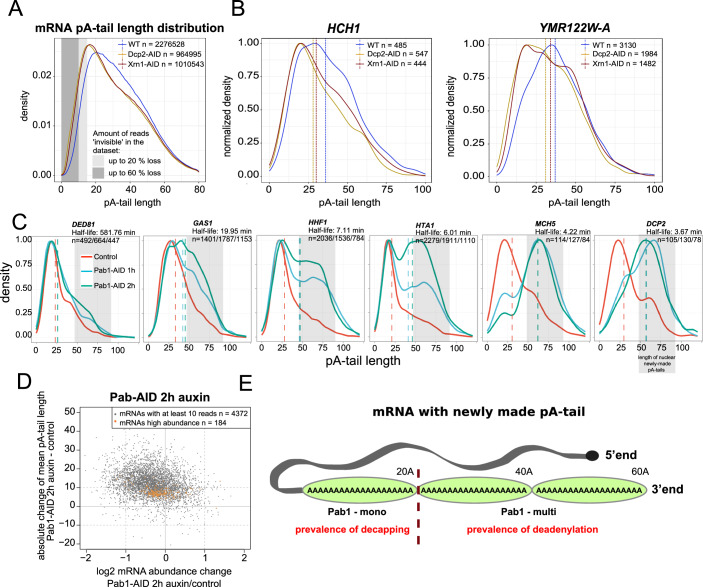
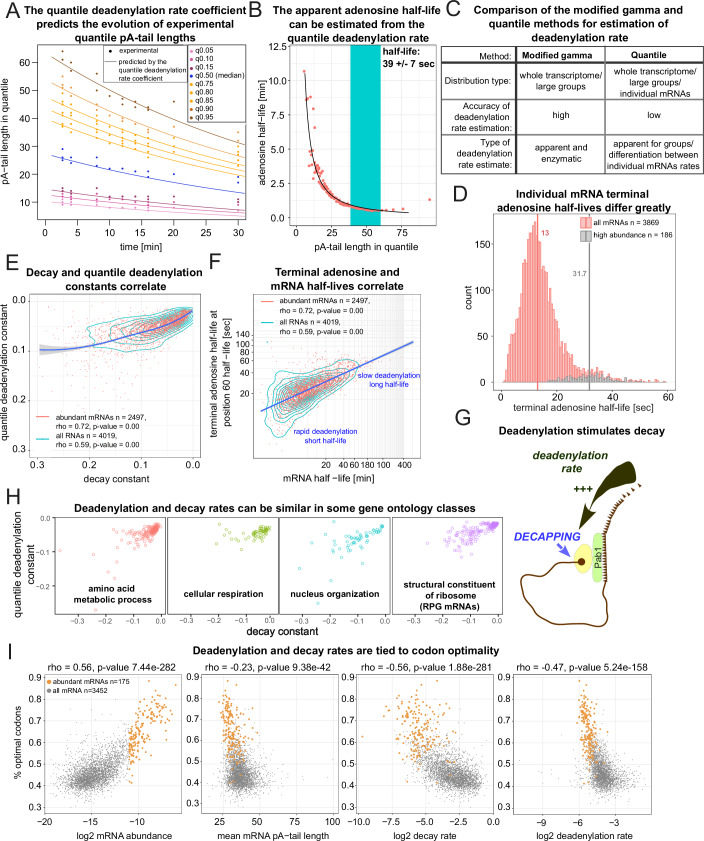
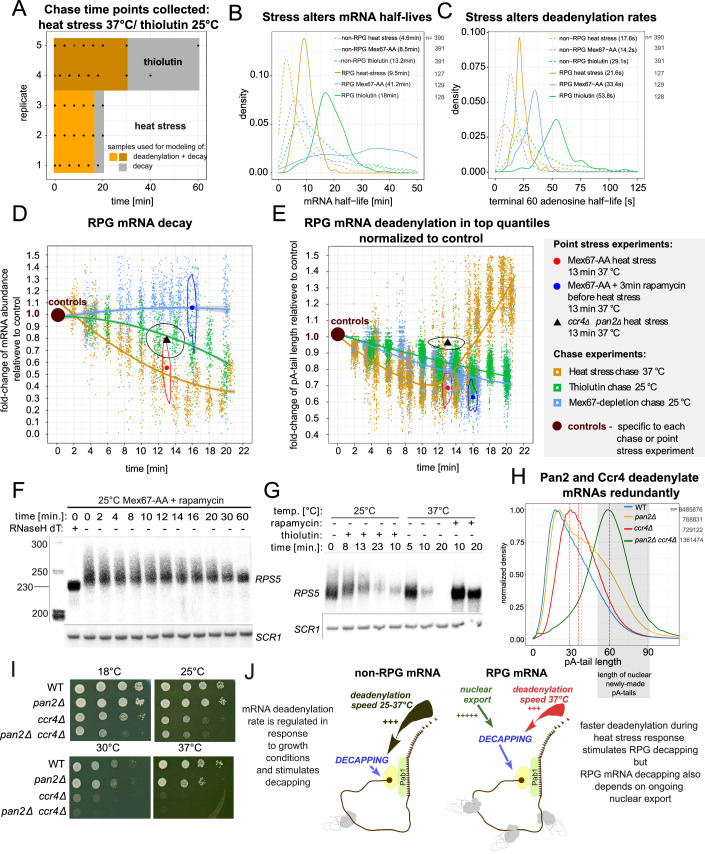
Complete cytoplasmic polyadenosine tail (polyA-tail) deadenylation is thought to be essential for initiating mRNA decapping and subsequent degradation. To investigate this prevalent model, we conducted direct RNA sequencing of S. cerevisiae mRNAs derived from chase experiments under steady-state and stress condition. Subsequently, we developed a numerical model based on a modified gamma distribution function, which estimated the transcriptomic deadenylation rate at 10 A/min. A simplified independent method, based on the delineation of quantile polyA-tail values, showed a correlation between the decay and deadenylation rates of individual mRNAs, which appeared consistent within functional transcript groups and associated with codon optimality. Notably, these rates varied during the stress response. Detailed analysis of ribosomal protein-coding mRNAs (RPG mRNAs), constituting 40% of the transcriptome, singled out this transcript group. While deadenylation and decay of RPG mRNAs accelerated under heat stress, their degradation could proceed even when deadenylation was blocked, depending entirely on ongoing nuclear export. Our findings support the general primary function of deadenylation in dictating the onset of decapping, while also demonstrating complex relations between these processes.
Keywords: Ccr4-NOT and Pan2/3 Deadenylases; Dcp2 Decapping and Xrn1 Degradation; ONT Nanopore Direct RNA Sequencing (DRS); Pab1; mRNA Deadenylation and Degradation.
© 2024. The Author(s).
Conflict of interest statement
Disclosure and competing interests statement. The authors declare no competing interests.
Figures






References
-
- Aibara S, Gordon JM, Riesterer AS, McLaughlin SH, Stewart M (2017) Structural basis for the dimerization of Nab2 generated by RNA binding provides insight into its contribution to both poly(A) tail length determination and transcript compaction in Saccharomyces cerevisiae. Nucleic Acids Res 45(3):1529–1538 - PMC - PubMed
-
- Albert R, Barabási AL (2002) Statistical mechanics of complex networks. Rev Mod Phys 74(1):47–97. 10.1103/RevModPhys.74.47
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
