

Pathogenic cryptic variants detectable through exome data reanalysis significantly increase the diagnostic yield in Joubert syndrome
- PMID: 39394465
- PMCID: PMC11711660
- DOI: 10.1038/s41431-024-01703-x
Pathogenic cryptic variants detectable through exome data reanalysis significantly increase the diagnostic yield in Joubert syndrome
Abstract
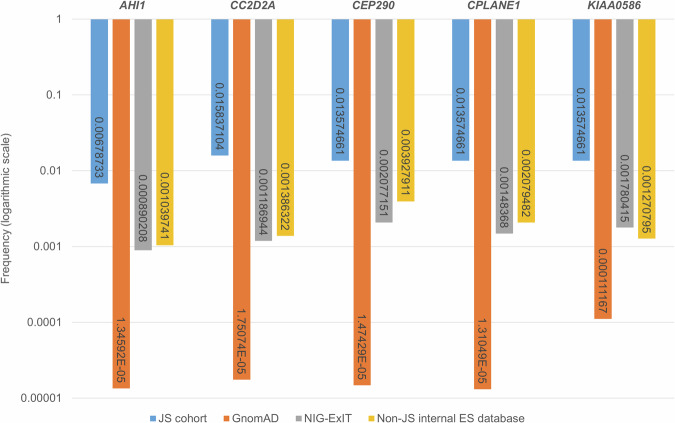
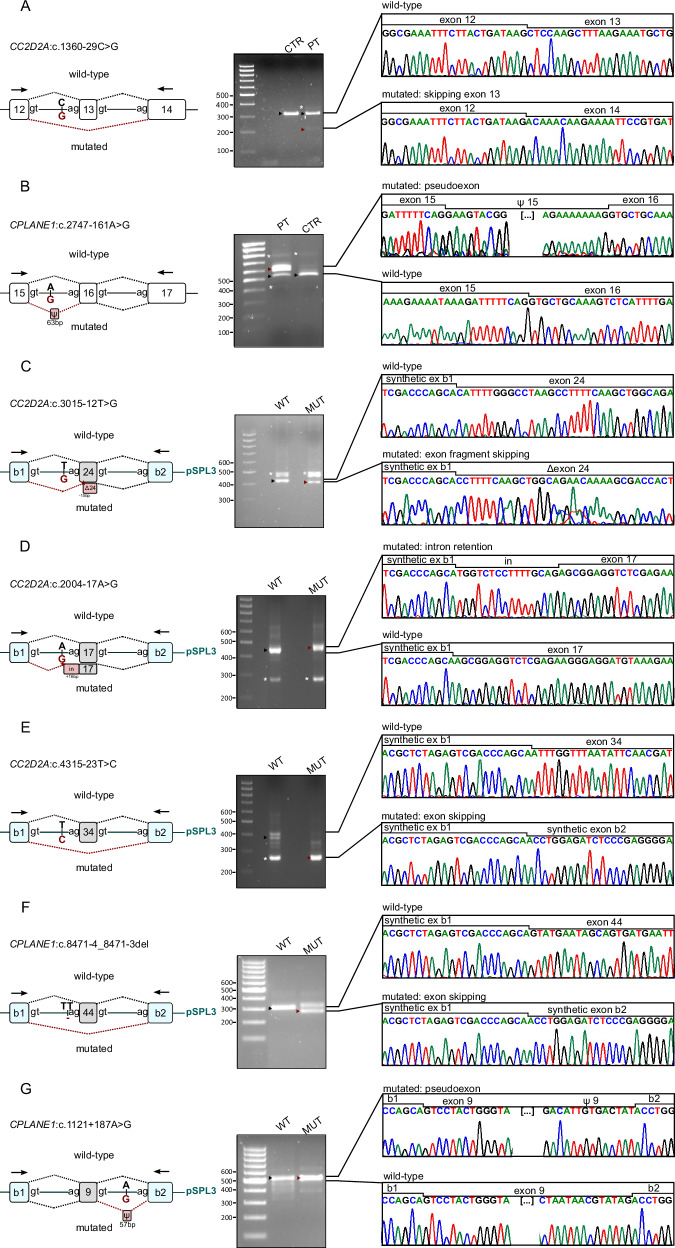
Joubert syndrome (JS) is a genetically heterogeneous neurodevelopmental ciliopathy. Despite exome sequencing (ES), several patients remain undiagnosed. This study aims to increase the diagnostic yield by uncovering cryptic variants through targeted ES reanalysis. We first focused on 26 patients in whom ES only disclosed heterozygous pathogenic coding variants in a JS gene. We reanalyzed raw ES data searching for copy number variants (CNVs) and intronic variants affecting splicing. We validated CNVs through real-time PCR or chromosomal microarray, and splicing variants through RT-PCR or minigenes. Cryptic variants were then searched in additional 44 ES-negative JS individuals. We identified cryptic "second hits" in 14 of 26 children (54%) and biallelic cryptic variants in 3 of 44 (7%), reaching a definite diagnosis in 17 of 70 (overall diagnostic gain 24%). We show that CNVs and intronic splicing variants are a common mutational mechanism in JS; more importantly, we demonstrate that a significant proportion of such variants can be disclosed simply through a focused reanalysis of available ES data, with a significantly increase of the diagnostic yield especially among patients previously found to carry heterozygous coding variants in the KIAA0586, CC2D2A and CPLANE1 genes.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests. Ethical approval: This research study adheres to the principles in the Declaration of Helsinki and was approved by the medical ethical committee of the University of Pavia (Nr. 20210017314 dated 13/05/2021). Written informed consent was obtained from parents or legal representatives of all the enrolled patients for clinical testing and publication of genetic and clinical data.
Figures
References
MeSH terms
Substances
Supplementary concepts
Grants and funding
- GGP20070/Fondazione Telethon (Telethon Foundation)
- Ricerca Finalizzata RF-2019-12369368/Ministero della Salute (Ministry of Health, Italy)
- RC 2023 line 1/Ministero della Salute (Ministry of Health, Italy)
- MNESYS (PE0000006)/Ministero dell'Istruzione, dell'Università e della Ricerca (Ministry of Education, University and Research)
LinkOut - more resources
Full Text Sources
Medical
